
LA DERMATOMIOSITIS
")
")
")
 en muslos, dermatomiositis")
DERMATOMIOSITIS CLÁSICA:
La dermatomiositis es una enfermedad clasificada como una colagenosis dentro del grupo de las enfermedades autoinmunes. Afecta principalmente la piel y los músculos. Clasificada dentro de las miopatías inflamatorias idiopáticas, siendo la característica distintiva de esta patología la inflamación de los músculos esqueléticos, y lesiones cutáneas distintivas.
Bohan y Peter en el año 1975, establecen 5 criterios con el objetivo de clasificar y diagnosticar la DERMATOMIOSITIS, de los cuales, 4 de ellos se referían a la clínica muscular: debilidad simétrica, proximal y progresiva, aumento de las enzimas musculares, electromiografía anormal y biopsia muscular anormal. El 5to criterio son las manifestaciones cutáneas compatibles, que es lo que diferencia esta patología de la POLIMIOSITIS. (PM)
Para esta fecha el término DERMATOMIOSITIS AMIOPÁTICA no se conocía ni estaba definido, hecho que ocurre en 1979 por el Dr., Pearson, lo cual explicaré mas adelante.
1.) SÍNTOMAS Y SIGNOS:
1.) Debilidad Muscular:
Afecta principalmente los músculos proximales, como los de los hombros, caderas y muslos, lo que puede dificultar actividades cotidianas como subir escaleras o levantar objetos. El paciente muestra debilidad para alzar los brazos, subir cuestas o escaleras y fatiga generalizada.
2.) Manifestaciones Cutáneas:
A.- Erupción en Heliotropo: Un rash violáceo en los párpados y alrededor de los ojos.
B.- Pápulas de Gottron: Lesiones cutáneas elevadas o planas de color violáceo o púrpura en las articulaciones, especialmente en los nudillos.
C.- Signo del Chal: Eritema o enrojecimiento que se extiende desde la parte posterior del cuello hasta la base del cuello.
B.- Eritema: Enrojecimiento generalizado, especialmente en áreas expuestas al sol.
D.- Signo de Holster: Eritema o enrojecimiento y edema que se presenta en las áreas laterales de los muslos, comúnmente llamado signo de la "pistolera", o "cartuchera".
2.) SÍNTOMAS SISTÉMICOS
Fatiga generalizada, fiebre, disminución de peso y malestar general pueden acompañar a la debilidad muscular y las lesiones cutáneas. Otros síntomas que pueden manifestarse: fenómeno de Raynaud, disfonía, y poliartritis inflamatoria no erosiva.
3.) DIAGNÓSTICO:
El diagnóstico de la dermatomiositis se basa en una combinación de hallazgos clínicos, pruebas de laboratorio biopsia cutánea , biopsia de músculo estriado y pruebas de laboratorio.
4.) LABORATORIO:
A.- Elevación de enzimas musculares: la creatina quinasa (CK),
Esta es la enzima más comúnmente asociada con lesiones musculares. En la dermatomiositis, los niveles de CK pueden elevarse significativamente, a menudo hasta 50 veces por encima del valor normal. Se calcula que entre el 80% y el 90% de los pacientes con miositis activa presentan elevaciones en esta enzima CK.
B.- Deshidrogenasa láctica (DHL):
Este indicador nos muestra el daño muscular. Cuando las fibras musculares se inflaman y destruyen, se liberan enzimas intracelulares, incluida la LDH, al torrente sanguíneo.
El valor normal es de 140 a 280 U/L dependiendo del laboratorio; en la dermatomiositis puede encontrarse hasta 1000 U/L, su valor depende del daño muscular.
C.- Aldolasa:
La aldolasa es una enzima que se utiliza como marcador para evaluar el daño muscular. Se considera que los niveles de aldolasa están elevados cuando superan el rango normal, que generalmente oscila entre 1.0 a 7.5 unidades por litro (U/L)
D.- Detección de autoanticuerpos específicos:
1.) Anticuerpos antinucleares ANA: 80% de los pacientes.
2.) Anticuerpos contra histidina-ARNt ligasa: (Anti-Jo-1 ANTI-PL-7) 30% de los pacientes.
3.) Anticuerpos anti-Mi-2.
5.) BIOPSIA MUSCULAR:
Se realiza para confirmar la presencia de infiltrados inflamatorios y cambios característicos asociados con la dermatomiositis, la fibra muscular por lo general es obtenida mediante un proceso de cirugía menor en el área deltoidea.
Histopatológicamente los hallazgos son: como característica distintiva la atrofia perifascicular, donde las fibras musculares en los bordes de las fascículos están atrofiadas, mientras que las fibras en el centro pueden estar conservadas; también hay una afectación vascular notable, con disminución de los capilares en áreas afectadas, lo que contribuye a la isquemia muscular.
6.) ELECTROMIOGRAFÍA (EMG):
Puede ayudar a evaluar la función muscular y detectar patrones típicos de daño muscular como : fibrilaciones (descargas eléctricas anormales) y Ondas Agudas Positivas; también descargas Repetitivas de Alta Frecuencia. La EMG ayuda al diagnóstico pero en un 10% de los casos da resultados normales.
7.) IMAGENOLOGÍA:
La Resonancia magnética (RM) se utiliza para evaluar el compromiso muscular y guiar las biopsias si es necesario.
8.) TRATAMIENTO:
A.- Corticoides:
Prednisona: Se utiliza como tratamiento inicial para controlar rápidamente los síntomas. La dosis suele ser alta al principio y luego se reduce gradualmente a medida que mejoran los síntomas.
Dosis: 60 mg y 100 mg al día. Esta dosis inicial tiene como objetivo controlar rápidamente la inflamación y prevenir el daño muscular durante las primeras semanas de tratamiento.
Dosis de Mantenimiento: Suelen oscilar entre 5 mg y 20 mg al día, dependiendo de la respuesta clínica del paciente y la severidad de los síntomas. Pudieran utilizarse 30 mg día y ajustarse según la evolución del paciente.
Las dosis se van disminuyendo de manera gradual, generalmente entre un 10% y un 20% cada 1 o 2 semanas, hasta alcanzar una dosis mínima que controle los síntomas.
Los corticoides pueden utilizarse en conjunción con otros medicamentos como la azatioprina y el metotrexato en esta patología.
B.- Inmunosupresores:
1.) Azatioprina: Ayuda a reducir la dosis de corticoides y controlar la inflamación muscular. dosis 1 a 3 mg/kg/día. mantenimiento 1-2 mg/kg/día.
2.) Metotrexato: Se usa para suprimir el sistema inmunológico y mejorar los síntomas.
La dosis inicial de metotrexato en adultos suele ser de 15 mg a 25 mg semanales. Algunos protocolos sugieren comenzar con una dosis más baja, como 7.5 mg a 10 mg semanales, y luego aumentar gradualmente.
Estas dosis se van ajustando de acuerdo a la respuesta del paciente pudiendo incrementarse en intervalos de 2.5 a 5 mg cada 2 a 4 semanas, según la respuesta clínica y los efectos secundarios, hasta alcanzar un máximo de 25 a 30 mg semanales.
3.) Micofenolato de mofetilo: Útil si hay afectación pulmonar.
La dosis inicial recomendada es de 1 a 2 g/día, dividida en dos tomas. Esto se traduce generalmente en 500 mg a 1 g dos veces al día, con una dosis máxima que no debe excederse de 2.5 g/día.
4.) Ciclofosfamida: Considerada en casos severos o resistentes al tratamiento.
La dosis típica es de 500 a 1000 mg/día, administrada cada 3 a 4 semanas. Este régimen se suele mantener durante 3 a 6 meses, dependiendo de la respuesta clínica.
Dosis Oral: La ciclofosfamida también puede utilizarse oralmente en dosis de aproximadamente 1-2 mg/kg/día.
5.) Terapias Biológicas:
1.) Rituximab: anticuerpo monoclonal utilizado en pacientes que no responden a tratamientos convencionales. Actúa reduciendo la cantidad de linfocitos B, disminuyendo así la actividad autoinmune.
La dosis clásica es de 1 g administrado por vía intravenosa, seguido de una segunda dosis de 1 g dos semanas después. Este esquema se puede repetir según la respuesta del paciente.
Para minimizar los efectos de la infusión se utilizan antihistamínicos y paracetamol.
6.) Medicamentos Antipalúdicos:
1.) Hidroxicloroquina: Se puede utilizar para tratar lesiones cutáneas persistentes.
La dosis habitual de hidroxicloroquina en adultos es de 5 a 6 mg/kg/día, administrada en una sola toma diaria, con un máximo de 400 mg al día, por lo general dividida en una dosis de 200 mg cada 12 horas.
La hidroxicloroquina puede usarse a largo plazo por unas 4 a 8 semanas evaluando siempre los efectos secundarios y la respuesta clínica del paciente.
7.) Terapias Complementarias:
1.) Fisioterapia: Para mejorar la fuerza y flexibilidad muscular.
2.) Terapia del habla: cuando hay afectación en los músculos de la deglución.
3.) Nutrición: En caso de problemas relacionados con la alimentación, se recomienda la asistencia por un dietista.
4.) Protectores solares: es indispensable proteger la piel del sol, para evitar el agravamiento de los síntomas cutáneos.
8.) Tratamientos Adicionales:
1.) Inmunoglobulina Intravenosa (IVIG): Inmunoglobulina G intravenosa. Tratamiento usado en casos severos para bloquear anticuerpos dañinos. Las más comúnmente utilizadas son la Privigen, Gammagard, y Octagam.
La inmunoglobulina intravenosa (IVIG) es un tratamiento que se utiliza en la dermatomiositis, especialmente en casos severos o refractarios a otros tratamientos; específicamente utilizada en casos donde encontramos:
1.) Afectación Muscular Grave: Primera línea de tratamiento en casos de miopatía grave o fulminante.
2.) Refractariedad a Corticoides: Cuando los pacientes no responden adecuadamente a los esteroides presentando empeoramiento de los síntomas, al reducir las dosis adecuadas.
3.) Manifestaciones Cutáneas Severas: lesiones cutáneas intensas y extensas, aun sin presencia de síntomas musculares.
Eficacia de su uso:
En algunos estudios se ha encontrado que la utilización de la inmunoglobulina G por vía intravenosa (IVIG) ha mejorado la sintomatología en un 79% de los casos tratados en comparación con un 44% del grupo placebo. Los mejores efectos se describen en mejoría de la fuerza muscular y síntomas neuromusculares.
La dosis típica de IVIG es de 2 g/kg de peso corporal, administrada cada 4 semanas. Este régimen debe ajustarse según la respuesta del paciente y la tolerancia al tratamiento.
Notas: Para que la efectividad del tratamiento sea a largo plazo el tratamiento debe ser repetitivo, otra consideración es que este tratamiento no debe ser usado como MONOTERAPIA, debe ser adyuvante de los tratamientos clásicos; y por último es un tratamiento costoso, lo cual puede ser un limitante para pacientes de bajos recursos o no asegurados.
Los efectos secundarios más comunes del uso de la IVIG son: Cefaleas, fiebre, náusea y eventos tromboembólicos.
Para ver más efectos secundarios de estos medicamentos léase la actualización de LUPUS ERITEMATOSO SISTÉMICO, TRATAMIENTO
DERMATOMIOSITIS AMIOPÁTICA:
En el año 1979, el médico Pearson es el primero en proponer el término "dermatomiositis amiopática" para describir pacientes que presentaban erupciones cutáneas típicas de dermatomiositis sin evidencia de debilidad muscular significativa.
Este término se utilizó para describir una variante de la enfermedad que constituye hoy día 2024, entre el 2% y el 18% de los casos de dermatomiositis, y que en este grupo entre el 6 y 60% de los casos tienden a desarrollar malignidades, específicamente tumores sólidos como cáncer de mama y genitourinarios. Entonces estaríamos hablando en estos casos de síndromes paraneoplásicos asociados a esta patología.
En el año 1993 los investigadores Euwer y Sontheimer propusieron criterios diagnósticos específicos para la dermatomiositis amiopática Clásica (DMA), los cuales son:
1.) Lesiones cutáneas características de dermatomiositis.
2.) Biopsia cutánea compatible con dermatomiositis.
3.) Ausencia de debilidad muscular proximal durante al menos dos años después del inicio de las lesiones cutáneas, hoy reducido este término a 6 meses.
4.) Sin elevación de enzimas musculares séricas dentro del mismo periodo (6 meses).
5.) Biopsia Muscular sin evidencias típicas de daño de la fibra muscular.
En este espectro se describió otra variante: la Dermatomiositis Hipoamiopatica: hecho que ocurre en el año 2002 por el médico Sontheimer.
Siendo considerado ese año 2002 la fecha donde quedan instaurados los términos definitivos de: Dermatomiositis amiopática clásica y Dermatomiositis hiopamiopatica.
Donde además de todos los criterios anteriores (1-4), en el criterio 5 sobre la biopsia muscular, se pueden observar cambios o rasgos de alteraciones. (en la hipoamiopatica)
Para el tratamiento de esta condición NO SE RECOMIENDA el uso de corticosteroides orales, el tratamiento es mas conservador, siendo la hidroxicloroquina y los esteroides tópicos los más recomendados. Sin embargo siendo una patología rara, es importante su reconocimiento por la asociación entre esta y el desarrollo de malignidades.
Saludos,,,
Dr. José Lapenta.
ENGLISH
CLASSIC DERMATOMYOSITIS:
Dermatomyositis is a disease classified as a collagenosis within the group of autoimmune diseases. It mainly affects the skin and muscles. Classified within idiopathic inflammatory myopathies, the distinctive characteristic of this pathology being inflammation of the skeletal muscles, and distinctive skin lesions.
HISTORY:
In 1975, Bohan and Peter established 5 criteria with the aim of classifying and diagnosing DERMATOMYOSITIS, of which 4 of them referred to the muscular symptoms: symmetrical, proximal and progressive weakness, increased muscle enzymes, abnormal electromyography and abnormal muscle biopsy. The 5th criterion is the compatible skin manifestations, which is what differentiates this pathology from POLYMYOSITIS. (PM)
At this time, the term AMYOPATHIC DERMATOMYOSITIS was not known or defined, a fact that occurred in 1979 by Dr. Pearson, which I will explain later.
1.) SYMPTOMS AND SIGNS:
1.) Muscle Weakness:
It mainly affects the proximal muscles, such as those of the shoulders, hips and thighs, which can make daily activities such as climbing stairs or lifting objects difficult. The patient shows weakness when raising the arms, climbing slopes or stairs and generalized fatigue.
2.) Skin Manifestations:
A.- Heliotrope rash: A purplish rash on the eyelids and around the eyes.
B.- Gottron's papules: Raised or flat, purplish or purple skin lesions on the joints, especially the knuckles.
C.- Chal's sign: Erythema or redness extending from the back of the neck to the base of the neck.
B.- Erythema: Generalized redness, especially in areas exposed to the sun.
D.- Holster's sign: Erythema or redness and edema that appears in the lateral areas of the thighs.
2.) SYSTEMIC SYMPTOMS:
Generalized fatigue, fever, weight loss, and malaise may accompany muscle weakness and skin lesions. Other symptoms that may occur: Raynaud's phenomenon, dysphonia, and non-erosive inflammatory polyarthritis.
3.) DIAGNOSIS:
The diagnosis of dermatomyositis is based on a combination of clinical findings, laboratory tests skin biopsy, striated muscle biopsy, and laboratory tests.
4.) LABORATORY:
A.- Elevation of muscle enzymes: creatine kinase (CK),
This is the enzyme most commonly associated with muscle lesions. In dermatomyositis, CK levels can be significantly elevated, often up to 50 times above the normal value. It is estimated that between 80% and 90% of patients with active myositis have elevations in this CK enzyme.
B.- Lactic dehydrogenase (LDH):
This indicator shows us muscle damage. When muscle fibers become inflamed and destroyed, intracellular enzymes, including LDH, are released into the bloodstream.
The normal value is 140 to 280 U/L depending on the laboratory; in dermatomyositis it can be found up to 1000 U/L, its value depends on the muscle damage.
C.- Aldolase:
Aldolase is an enzyme used as a marker to evaluate muscle damage. Aldolase levels are considered elevated when they exceed the normal range, which generally ranges from 1.0 to 7.5 units per liter (U/L)
D.- Detection of specific autoantibodies:
1.) Antinuclear antibodies ANA: 80% of patients.
2.) Antibodies against histidine-tRNA ligase: (Anti-Jo-1 ANTI-PL-7) 30% of patients.
3.) Anti-Mi-2 antibodies.
5.) MUSCLE BIOPSY:
It is performed to confirm the presence of inflammatory infiltrates and characteristic changes associated with dermatomyositis, the muscle fiber is usually obtained through a minor surgical process in the deltoid area.
Histopathologically, the findings are: as a distinctive characteristic, perifascicular atrophy, where the muscle fibers at the edges of the fascicles are atrophied, while the fibers in the center may be preserved; there is also a notable vascular affectation, with a decrease in capillaries in affected areas, which contributes to muscle ischemia.
6.) ELECTROMYOGRAPHY (EMG):
It can help evaluate muscle function and detect typical patterns of muscle damage such as: fibrillations (abnormal electrical discharges) and Positive Sharp Waves; also Repetitive High Frequency discharges. EMG helps with diagnosis but in 10% of cases it gives normal results.
7.) IMAGING:
Magnetic Resonance Imaging (MRI) is used to evaluate muscle involvement and guide biopsies if necessary.
8.) TREATMENT:
A.- Corticosteroids:
Prednisone: It is used as an initial treatment to quickly control symptoms. The dose is usually high at first and then gradually reduced as symptoms improve.
Dose: 60 mg and 100 mg daily. This initial dose is intended to quickly control inflammation and prevent muscle damage during the first weeks of treatment.
Maintenance dose: Usually ranges from 5 mg to 20 mg per day, depending on the patient's clinical response and the severity of symptoms. 30 mg per day may be used and adjusted according to the patient's progress.
Doses are gradually decreased, generally by 10% to 20% every 1 or 2 weeks, until reaching a minimum dose that controls symptoms.
Corticosteroids can be used in conjunction with other medications such as azathioprine and methotrexate in this pathology.
B.- Immunosuppressants:
1.) Azathioprine: Helps reduce the dose of corticosteroids and control muscle inflammation. dose 1 to 3 mg/kg/day. maintenance 1-2 mg/kg/day.
2.) Methotrexate: Used to suppress the immune system and improve symptoms.
The initial dose of methotrexate in adults is usually 15 mg to 25 mg weekly. Some protocols They suggest starting with a lower dose, such as 7.5 mg to 10 mg weekly, and then gradually increasing.
These doses are adjusted according to the patient's response and can be increased in 2.5 to 5 mg increments every 2 to 4 weeks, depending on clinical response and side effects, up to a maximum of 25 to 30 mg weekly.
3.) Mycophenolate mofetil: Useful if there is lung involvement.
The recommended starting dose is 1 to 2 g/day, divided into two doses. This generally translates to 500 mg to 1 g twice a day, with a maximum dose not to exceed 2.5 g/day.
4.) Cyclophosphamide: Considered in severe or treatment-resistant cases.
The typical dose is 500 to 1000 mg/day, administered every 3 to 4 weeks. This regimen is usually maintained for 3 to 6 months, depending on the clinical response.
Oral Dosage: Cyclophosphamide can also be used orally at doses of approximately 1-2 mg/kg/day.
5.) Biological Therapies:
1.) Rituximab: monoclonal antibody used in patients who do not respond to conventional treatments. It works by reducing the number of B lymphocytes, thus decreasing autoimmune activity.
The classic dose is 1 g administered intravenously, followed by a second dose of 1 g two weeks later. This regimen can be repeated depending on the patient's response.
Antihistamines and paracetamol are used to minimize the effects of the infusion.
6.) Antimalarial Drugs:
1.) Hydroxychloroquine: Can be used to treat persistent skin lesions.
The usual dose of hydroxychloroquine in adults is 5 to 6 mg/kg/day, administered in a single daily dose, with a maximum of 400 mg per day, usually divided into a dose of 200 mg every 12 hours.
Hydroxychloroquine can be used long-term for about 4 to 8 weeks, always evaluating the side effects and the patient's clinical response.
7.) Complementary Therapies:
1.) Physiotherapy: To improve muscle strength and flexibility.
2.) Speech therapy: when swallowing muscles are affected.
3.) Nutrition: In case of problems related to food, assistance from a dietician is recommended.
4.) Sunscreen: It is essential to protect the skin from the sun, to avoid the worsening of skin symptoms.
8.) Additional Treatments:
1.) Intravenous Immunoglobulin (IVIG): Intravenous immunoglobulin G. Treatment used in severe cases to block harmful antibodies. The most commonly used are Privigen, Gammagard, and Octagam.
Intravenous immunoglobulin (IVIG) is a treatment used in dermatomyositis, especially in severe cases or refractory to other treatments; specifically used in cases where we find:
1.) Severe Muscle Involvement: First line of treatment in cases of severe or fulminant myopathy.
2.) Refractoriness to Corticosteroids: When patients do not respond adequately to steroids presenting worsening of symptoms, when reducing the appropriate doses.
3.) Severe Cutaneous Manifestations: intense and extensive skin lesions, even without the presence of muscular symptoms.
Efficacy of its use:
In some studies, it has been found that the use of intravenous immunoglobulin G (IVIG) has improved symptoms in 79% of treated cases compared to 44% of the placebo group. The best effects are described in improved muscle strength and neuromuscular symptoms.
The typical dose of IVIG is 2 g/kg of body weight, administered every 4 weeks. This regimen should be adjusted according to the patient's response and tolerance to treatment.
Notes: For the treatment to be effective in the long term, the treatment must be repeated. Another consideration is that this treatment should not be used as MONOTHERAPY, it should be adjuvant to classic treatments; And lastly, it is an expensive treatment, which can be a limitation for low-income or uninsured patients.
The most common side effects of IVIG use are: Headaches, fever, nausea and thromboembolic events.
To see more side effects of these medications, read the update on SYSTEMIC LUPUS ERYTHEMATOSUS, TREATMENT
AMYOPATHIC DERMATOMYOSITIS:
In 1979, Dr. Pearson was the first to propose the term "amyopathic dermatomyositis" to describe patients who presented typical skin rashes of dermatomyositis without evidence of significant muscle weakness.
This term was used to describe a variant of the disease that today constitutes between 2% and 18% of dermatomyositis cases, and that in this group between 6 and 60% of cases tend to develop malignancies, specifically solid tumors such as breast and genitourinary cancer. In these cases, we would then be talking about paraneoplastic syndromes associated with this pathology.
In 1993, researchers Euwer and Sontheimer proposed specific diagnostic criteria for Classical Amyopathic Dermatomyositis (AMD), which are:
1.) Skin lesions characteristic of dermatomyositis.
2.) Skin biopsy compatible with dermatomyositis.
3.) Absence of proximal muscle weakness for at least two years after the onset of skin lesions, now reduced to 6 months.
4.) No elevation of serum muscle enzymes within the same period (6 months).
5.) Muscle biopsy without typical evidence of muscle fiber damage.
Another variant was described in this spectrum: Hypoamyopathic Dermatomyositis: a fact that occurred in 2002 by the doctor Sontheimer.
That year 2002 was considered the date when the definitive terms of: Classical amyopathic dermatomyositis and hypoamyopathic dermatomyositis were established.
Where, in addition to all the previous criteria (1-4), in criterion 5 on the muscle biopsy, changes or signs of alterations can be observed (in hypoamyopathic).
For the treatment of this condition, the use of oral corticosteroids is NOT RECOMMENDED, the treatment is more conservative, with hydroxychloroquine and topical steroids being the most recommended. However, being a rare pathology, its recognition is important due to the association between it and the development of malignancies.
Greetings...
Dr. José Lapenta R.
EDITORIAL ESPANOL:
====================
Hola Amigos DERMAGICOS,,, el tema de hoy, LA DERMATOMIOSITIS,, otra enfermedad del tejido conectivo cuyo tratamiento representa un verdadero reto, asociada a malignidad en algunos casos y de pronóstico reservado en otros tantos. Pienso que estas 38 referencias son bastante ilustrativas.
Les recuerdo que DERMAGIC/EXPRESS esta disponible en CD-ROM, !!!Desde Octubre 1998 hasta Mayo 1999, más de 1.000 referencias Bibliográficas actualizadas repartidas en unos 50 temas,,,
PRÓXIMA EDICIÓN: ESCLEROSIS SISTÉMICA PROGRESIVA,
Saludos,,,
Dr. José Lapenta R.,,,
EDITORIAL ENGLISH:
===================
Hello DERMAGIC friends, today's topic, THE DERMATOMYOSITIS, another illness of the connective tissue whose treatment represents a true challenge, associated to malignancy in some cases, and in others of uncertain future. I think that these 38 references are quite illustrative.
NEXT EDITIONS: SYSTEMIC SCLEROSIS (SCLERODERMA)
Greetings,,,
Dr. José Lapenta R.
=====================================================================
DERMAGIC/EXPRESS(55)
=====================================================================
DERMATOMIOSITIS / DERMATOMYOSITIS
=====================================================================
A.) Cutaneous Dermatomyositis in the Era of Biologicals.
B.) Clinical Presentation and Evaluation of Dermatomyositis.
C.) The Clinical Features, Diagnosis and Classification of Dermatomyositis.
E.) Dermatomyositis and Juvenile Dermatomyositis.
J.) Amyopathic Dermatomyositis: Retrospective Review of 37 Cases.
K.) Clinical, Histologic and Prognostic Features of Clinically Amyopathic Dermatomyositis.
M.) Amyopathic Dermatomyositis: Definitions, Diagnosis, and Management.
N.) Pharmacological Strategies in Dermatomyositis: Current Treatments and Future Directions.
O.) Update on the Pharmacological Treatment of Adult Myositis.
P.) Up-to-Date Treatment and Management of Myositis.
Q.) Current and New Targets for Treating Myositis.
R.) New Therapies in Anti-Mda5 Antibody-Positive Dermatomyositis.
S.) Novel Therapeutic Targets in Dermatomyositis.
1.) [Cyclophosphamide pulse therapy for refractory rheumatic diseases]
2.) Fulminant Pneumocystis carinii pneumonia in 4 patients with
dermatomyositis.
3.) A retrospective study of 13 Oriental children with juvenile
dermatomyositis.
4.) Pulsed intravenous methylprednisolone therapy for inflammatory
myopathies:
evaluation of the effect by comparing two consecutive biopsies from the
same muscle.
5.) [Generalized Mycobacterium avium-intracellulare infection due to
immunosuppressive
therapy of paraneoplastic dermatomyositis]
6.) Occurrence of polymyositis in the county of Gavleborg, Sweden.
7.) Cyclosporin a therapy in refractory juvenile dermatomyositis.
Experience and
longterm followup of 6 cases.
8.) Cyclosporine treatment for polymyositis/dermatomyositis: is it possible
to rescue
the deteriorating cases with interstitial pneumonitis?
9.) Progressive multifocal leukoencephalopathy during cyclosporine
treatment. A case
report.
10.) Update on therapy for refractory dermatomyositis and polymyositis.
11.) Partial lipodystrophy associated with juvenile dermatomyositis: report
of two
cases.
12.) Lesions resembling malignant atrophic papulosis in a patient with
dermatomyositis.
13.) Low-dose methotrexate administered weekly is an effective
corticosteroid-sparing
agent for the treatment of the cutaneous manifestations of dermatomyositis.
14.) Serum concentrations of carboxyterminal propeptide of type 1 procollogen
and tissue inhibitor of metalloproteinase 1 in patients with dermatomyositis.
15.) Dermatomyositis: a dermatology-based case series.
16.) Microvascular changes in skeletal muscle in idiopathic inflammatory
myopathy.
17.) Risk of cancer in patients with dermatomyositis or polymyositis. A
population-based study.
18.) Ovarian malignancy in patients with dermatomyositis and polymyositis: a
retrospective analysis of fourteen cases.
19.) Breast cancer and second primary ovarian cancer in dermatomyositis.
20.) [Vascular manifestations of dermatomyositis and polymyositis. Clinical,
capillaroscopic and histological aspects]
21.) Long-term prognosis of 69 patients with dermatomyositis or polymyositis.
22.) Dermatomyositis and malignancy.
23.) Dermatomyositis. Disease associations and an evaluation of screening
investigations for malignancy.
24.) [Dermatomyositis complicated by sarcoidosis]
25.) [Prognostic factors and predictive signs of malignancy in adult
dermatomyositis]
26.) Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed.
27.) Cutaneous changes of dermatomyositis in patients with normal muscle
enzymes: dermatomyositis sine myositis?
28.) Association between bovine collagen dermal implants and a dermatomyositis
or a polymyositis-like syndrome [see comments]
29.) MR imaging in amyopathic dermatomyositis.
30.) Case Report: amyopathic dermatomyositis associated with transformed
malignant lymphoma.
31.) Malignancy in adult dermatomyositis.
32.) Treacher-Collins syndrome and co-existing dermatomyositis.
33.) [A clinical analysis of cutaneous type dermatomyositis]
34.) Dermatomyositis with a pityriasis rubra pilaris-like eruption: a
little-known distinctive cutaneous manifestation of dermatomyositis.
35.) HCV and dermatomyositis: report of 5 cases of dermatomyositis in patients
with HCV infection.
36.) [Dermatomyositis and cancer. A retrospective study]
37.) Amyopathic dermatomyositis (dermatomyositis sin´e myositis). Presentation
of six new cases and review of the literature [see comments]
38.) Epstein-Barr virus strain type and latent membrane protein 1 gene
deletions in
lymphomas in patients with rheumatic diseases.
=====================================================================
=====================================================================
1.) [Cyclophosphamide pulse therapy for refractory rheumatic diseases]
=====================================================================
Tateishi M; Taniguchi A; Moriguchi M; Hara M; Kashiwazaki S
Institute of Rheumatology, Tokyo women's Medical College.
Nihon Rinsho Meneki Gakkai Kaishi (JAPAN) Jun 1997 20 (3) p152-8
Language: JAPANESE Summary Language: ENGLISH
JOURNAL ARTICLE English Abstract
Journal Announcement: 9711
Subfile: INDEX MEDICUS
We studied the efficacy of cyclophosphamide pulse therapy (CYP) for
refractory
rheumatic diseases except for lupus nephritis. Thirty-five patients were
included in
all, that is 9 patients with systemic lupus erythematosus (SLE), 10 with
rheumatoid
arthritis (RA), 7 with polymyositis/dermatomyositis (PM/DM), 2 with
progressive
systemic sclerosis (PSS), 2 with anti-phospholipid antibody syndrome (APS),
1 with
mixed connective tissue disease (MCTD), 1 with juvenile rheumatoid
arthritis (JRA), 1
with Behcet's disease (BD), 1 with Wegener's granulomatosis (WG) and 1 with
allergic
granulomatous angiitis (AGA). Moderate to marked improvement was noted in
4 patients
with SLE (2 with CNS-lupus and 2 with vasculitis), 7 with RA (2 with
interstitial
pneumonia:IP, 2 with vasculitis and 2 with refractory arthritis), 4 with
PM/DM (2
with IP and 2 with refractory myositis), 1 PSS with IP, 2 APS with
thrombocytopenia,
1 JRA with vasculitis and BD with CNS disturbance. On the other hand,
adverse
reactions were observed in 9 out of 35 patients (25.8%). CYP should apply
in the
treatment for such refractory rheumatic diseases as CNS disturbance,
vasculitis, IP
and autoimmune thrombocytopenia.
=====================================================================
2.) Fulminant Pneumocystis carinii pneumonia in 4 patients with
dermatomyositis.
=====================================================================
Bachelez H; Schremmer B; Cadranel J; Mouly F; Sarfati C; Agbalika F;
Schlemmer B;
Mayaud CM; Dubertret L
Institut de Recherche sur la Peau, Hopital Saint-Louis, Paris, France.
Arch Intern Med (UNITED STATES) Jul 14 1997 157 (13) p1501-3 ISSN:
0003-9926
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9710
Between 1989 and 1996, 4 cases of Pneumocystis carinii pneumonia (PCP) were
observed in patients seronegative for the human immunodeficiency virus who
were
receiving corticosteroid therapy for dermatomyositis in our institution.
These cases
were considered unusual in light of the short delay of their onset after
initiation
of immunosuppressive therapy and their fulminant course: 3 of these
patients died of
PCP occurring during the first month of treatment with prednisone. In all
4 patients
lymphopenia was observed before the initiation of corticosteroid treatment
and low
CD4 and CD8 cell counts were evident at the time of PCP. These
observations support
the view of an increase in both the severity and incidence of PCP in
patients without
human immunodeficiency virus infection and question the need for a primary
prophylaxis in patients with connective tissue diseases receiving high-dose
corticosteroid therapy.
=====================================================================
3.) A retrospective study of 13 Oriental children with juvenile
dermatomyositis.
=====================================================================
See Y; Giam YC; Chng HH
Department of Paediatrics, Alexandra Hospital, Singapore.
Ann Acad Med Singapore (SINGAPORE) Mar 1997 26 (2) p210-4 ISSN:
0304-4602
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9710
This is a retrospective study of 13 Oriental children with juvenile
dermatomyositis
(JDM). We reviewed data found in the hospital records of children
diagnosed to have
definite (n = 4), probable (n = 7) and possible (n = 2) JDM who presented
over a 10-
year period at 4 centres in Singapore and compared our results with the
experience of
others. We found an overall female preponderance (female to male ratio of
3.3:1) but
an equal sex ratio in children below 5 years of age. The majority (92%) had
insidious onset and good outcome. Diagnosis was often delayed because of the
insidious onset, and because weakness occurred late, was mild or absent.
Only one
child had an acute presentation and refractory course. She died despite
aggressive
therapy. Clinical features, complications and mainstay medication used
were similar
to Western studies.
=====================================================================
4.) Pulsed intravenous methylprednisolone therapy for inflammatory
myopathies:
evaluation of the effect by comparing two consecutive biopsies from the
same muscle.
=====================================================================
Matsubara S; Hirai S; Sawa Y
Department of Neurology, Tokyo Metropolitan Neurological Hospital, Japan. t-
mtbrs@tmin.ac.jp
J Neuroimmunol (NETHERLANDS) Jun 1997 76 (1-2) p75-80 ISSN: 0165-5728
Language: ENGLISH
To examine the effect of pulsed intravenous methylprednisolone (IVMP) on
polymositis (PM) and dermatomyositis (DM), two biopsies were taken from the
same
muscle before and after IVMP in one case each of PM and DM. After IVMP,
numerous
muscle fibers were seen undergoing regeneration. Among all subsets of
infiltrating
cells that decreased in number, macrophages in the perivascular area
decreased
markedly. Also conspicuous was decrease of CD8+ cells in the endomysium in
PM and
that of B-cells in the perivascular area in DM. Among infiltrating cells,
frequency
of those which expressed the signal transducers and activators of
transcription 1
(STAT-1) fell considerably, but remained still abnormally high. Regions on
the
endothelial cells of blood vessels that expressed MHC antigens and
intercellular
adhesion molecules 1 (ICAM-1) also decreased but remained more widespread
than normal.
These results agreed with the favorable clinical effect of IVMP, but showed
that many
of the immunological parameters remained abnormal.
=====================================================================
5.) [Generalized Mycobacterium avium-intracellulare infection due to
immunosuppressive
therapy of paraneoplastic dermatomyositis]
=====================================================================
Generalisierte Mycobacterium avium-intracellulare Infektion infolge
immunsuppressiver Therapie einer paraneoplastischen Dermatomyositis.
Schaller M; Korting HC; Meurer M; Schirren CG
Dermatologische Klinik und Poliklinik, Ludwig-Maximilians-Universitat,
Munchen.
Hautarzt (GERMANY) Feb 1997 48 (2) p118-21 ISSN: 0017-8470
Language: GERMAN Summary Language: ENGLISH
Document Type:
JOURNAL ARTICLE English Abstract
Subfile: INDEX MEDICUS
The ubiquitous Mycobacterium avium-intracellulare (MAI) is the most
frequent cause
of disseminated atypical mycobacteriosis in AIDS patients. MAI infections
may
develop in patients with other acquired immune defects, such as connective
tissue
disorders. In adults, the gastrointestinal and respiratory systems are most
frequently affected. We report a patient with dermatomyositis receiving
immunosuppressive therapy in whom only the skin and the skeletal system
were affected
by MAI. Because it presented with polymyositis-like symptoms, the
infection was
initially not identified and treated. The MAI was cultured from a
periarticular
joint effusion from the right upper arm and from venous blood, as well as
identified
histologically in lesional skin. Resistance to antibiotics developed most
likely
because the patient failed to take oral antibiotics regularly. Because of
an acute
exacerbation of the tumor-associated dermatomyositis, immunosuppressive
therapy was
initiated, while the tuberculostatic therapy was continued. Using these
therapies
both diseases markedly improved. In patients with connective tissue
disorders
receiving longterm immunosuppressive therapy, especially when changes in
symptoms and
signs are observed, opportunistic infections such as MAI should be
considered and
included in the differential diagnosis.
=====================================================================
6.) Occurrence of polymyositis in the county of Gavleborg, Sweden.
=====================================================================
Weitoft T
Department of Internal Medicine, Lanssjukhuset Gavle, Sweden.
Scand J Rheumatol (NORWAY) 1997 26 (2) p104-6 ISSN: 0300-9742
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9707
Subfile: INDEX MEDICUS
Through register research 21 new cases (10 male, 11 female) of
polymyositis/dermatomyositis during the period 1984-1993 in the county of
Gavleborg,
Sweden, were identified. The case records were studied retrospectively.
Inclusion
body myositis was found in three cases and overlap syndrome in seven cases.
Three
patients had associated malignant disease. The annual incidence was
estimated to 7.6
cases/million people. The clinical features are described. 19 patients
were given
steroid treatment, and azathioprin was the most used additive
immunosuppressive drug.
All patients could be followed for at least two years, and during this
period three
patients died (all of cancer disease). Remission and withdrawal of
medication were
achieved in five cases. The results of the study corresponds to previous
investigations with similar design.
=====================================================================
7.) Cyclosporin a therapy in refractory juvenile dermatomyositis.
Experience and
longterm followup of 6 cases.
=====================================================================
Zeller V; Cohen P; Prieur AM; Guillevin L
Department of Internal Medicine, Hopital Avicenne, Universite Paris-Nord,
Bobigny,
France.
J Rheumatol (CANADA) Aug 1996 23 (8) p1424-7 ISSN: 0315-162X
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9707
Subfile: INDEX MEDICUS
OBJECTIVE: Cyclosporin A (CyA) has been reported to be an effective
treatment of
adult polymyositis (PM) and dermatomyositis (DM) refractory to
corticosteroid therapy.
We aimed to determine the effectiveness of CyA in the treatment of juvenile
dermatomyositis (JDM) refractory to corticosteroids and cytotoxic agents.
METHODS:
Retrospective study of 6 patients with refractory JDM based on medical
charts.
RESULTS: Better clinical control of the disease was obtained in all cases and
corticosteroid doses could be markedly decreased or stopped. 4 patients
relapsed, 3
after discontinuation of CyA, but recovered after its reinitiation. Trough
CyA
levels were maintained between 51 and 247 ng/ml, and side effects were rare
and minor
during a mean followup of 51.5 months. CONCLUSION: CyA seems to be an
effective
treatment of JDM and should be considered in case of severe or refractory
disease.
=====================================================================
8.) Cyclosporine treatment for polymyositis/dermatomyositis: is it possible
to rescue
the deteriorating cases with interstitial pneumonitis?
=====================================================================
Maeda K; Kimura R; Komuta K; Igarashi T
Second Department of Internal Medicine, Osaka Teishin Hospital, Japan.
Scand J Rheumatol (NORWAY) 1997 26 (1) p24-9 ISSN: 0300-9742
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Polymyositis and dermatomyositis are inflammatory muscular diseases of
unknown
etiology which have interstitial pneumonitis as a serious complication.
From 1987 to
1994, we used cyclosporine to treat 14 polymyositis/dermatomyositis
patients (8 with
and 6 without interstitial pneumonitis). In combination with prednisolone,
cyclosporine was either added to or substituted for conventional cytotoxic
drugs
(azathioprine, cyclophosphamide and methotrexate). Cyclosporine was
effective
against both myositis and interstitial pneumonitis when used early in the
course of
the disease.
=====================================================================
9.) Progressive multifocal leukoencephalopathy during cyclosporine
treatment. A case
report.
=====================================================================
Gentile S; Sacerdote I; Roccatello D; Giordana MT
Divisione di Neurologia, Ospedale Giovanni Bosco, Torino, Italy.
Ital J Neurol Sci (ITALY) Oct 1996 17 (5) p363-6 ISSN: 0392-0461
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW OF REPORTED CASES
Journal Announcement: 9705
Subfile: INDEX MEDICUS
A 45-year-old patient suffering from chronic renal insufficiency developed
dermatopolymyositis. Since the patient did not respond to treatment with
high doses
of prednisone and immunoglobulin, concomitant cyclosporine A was added.
Four months
later, worsening signs of bilateral pyramidal disorder and an altered state
of
consciousness appeared. Serial computed tomography (CT) and magnetic
resonance
imaging (MRI) revealed multiple alterations of the cerebral white matter.
Cyclosporine was then discontinued. One month later, exitus occurred.
Microscopic
examination of the brain showed diffuse tumescence; histological
examination revealed
perivascular and diffuse lymphomonocyte infiltrations, areas of
demyelination,
astrocytes with bizarre nuclei, and oligodendrocytes with enlarged nuclei
due to
hyperchromatic inclusion. Morphological examination confirmed the presence
of
intranuclear icosahedral viral bodies. Progressive multifocal
leukoencephalopathy
was diagnosed. The literature contains only one report of an analogous
case observed
during a course of cyclosporine treatment for Wegener's granulomatosis. (20
References)
=====================================================================
10.) Update on therapy for refractory dermatomyositis and polymyositis.
=====================================================================
Villalba L; Adams EM
National Institutes of Health, National Institute of Arthritis and
Musculoskeletal
and Skin Diseases, Arthritis and Rheumatism Branch, Bethesda, MD
20892-1820, USA.
Curr Opin Rheumatol (UNITED STATES) Nov 1996 8 (6) p544-51 ISSN:
1040-8711
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9705
Subfile: INDEX MEDICUS
Evaluation of new therapies for the inflammatory myopathies is
complicated by the
heterogeneity of these syndromes as well as by the lack of internationally
accepted
definitions of disease categories and assessments of disease activity and
chronicity.
This review covers our opinion of therapies and emphasizes the need for an
early
rehabilitation evaluation for these patients. Oral corticosteroids are the
first
line of therapy for the inflammatory myopathies, but because of their side
effects
and the existence of a subset of patients in whom disease is controlled
only with
high-dose corticosteroids, we recommend considering the early use of a
second-line
immunomodulating agents or pulse intravenous methylprednisolone. A stepwise
progression of therapies is suggested for patients who have increasing muscle
weakness resulting from active disease. (61 References)
=====================================================================
11.) Partial lipodystrophy associated with juvenile dermatomyositis: report
of two
cases.
=====================================================================
Quecedo E; Febrer I; Serrano G; Martinez-Aparicio A; Aliaga A
Department of Dermatology, Hospital General Universitario, Valencia, Spain.
Pediatr Dermatol (UNITED STATES) Nov-Dec 1996 13 (6) p477-82 ISSN:
0736-8046
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9705
A 27-year-old woman and a 13-year-old girl diagnosed with juvenile
dermatomyositis
in childhood developed clinical findings of partial lipodystrophy 10 years
after
diagnosis. Exhaustive clinical and laboratory examinations showed an
association
with other abnormalities: hypertrichosis, steatohepatitis, and an abnormal
insulin
response to the glucose loading test in the first patient. Hypertrichosis,
steatohepatitis, insulin-resistant diabetes mellitus, and acanthosis
nigricans were
observed in the second patient. Renal function was normal in both patients.
Although a localized form of lipodystrophy has been reported associated with
connective tissue disease (connective tissue lipoatrophy), the partial form
has been
infrequently described in association with juvenile dermatomyositis.
=====================================================================
12.) Lesions resembling malignant atrophic papulosis in a patient with
dermatomyositis.
=====================================================================
Tsao H; Busam K; Barnhill RL; Haynes HA
Department of Dermatology, Harvard Medical School, Brookline, MA, USA.
J Am Acad Dermatol (UNITED STATES) Feb 1997 36 (2 Pt 2) p317-9 ISSN:
0190-9622
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9705
Subfile: INDEX MEDICUS
Many consider porcelain white atrophic papules as pathognomonic for
malignant
atrophic papulosis (MAP), or Degos' disease. During the past three
decades, five
patients with a collagen vascular disease have been reported to have
MAP-like lesions
as a manifestation of their underlying illness. We describe a patient with
dermatomyositis who had porcelain-white atrophic papules resembling malignant
atrophic papulosis.
=====================================================================
13.) Low-dose methotrexate administered weekly is an effective
corticosteroid-sparing
agent for the treatment of the cutaneous manifestations of dermatomyositis.
=====================================================================
Kasteler JS; Callen JP
Division of Dermatology, University of Louisville, KY, USA.
J Am Acad Dermatol (UNITED STATES) Jan 1997 36 (1) p67-71 ISSN:
0190-9622
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9704
BACKGROUND: The cutaneous manifestations of dermatomyositis can be the most
prominent finding and are often difficult to treat. OBJECTIVE: Our purpose
was to
determine whether low-dose methotrexate administered weekly in combination
with other
systemic therapies or as a sole systemic agent is effective in the
treatment of the
cutaneous disease in patients with dermatomyositis. METHODS: We reviewed
the records
of 13 patients who received oral methotrexate in doses ranging from 2.5 to
30 mg
weekly. Their skin lesions had not been completely responsive to
sunscreens, topical
corticosteroids, oral prednisone, oral antimalarial therapy, and, in one
patient
each, chlorambucil and azathioprine. RESULTS: At the end of the study
period, 4 of
these 13 patients were free of all cutaneous manifestations of
dermatomyositis, and
another four had almost complete clearing. In the remaining five patients,
methotrexate induced moderate clearing of their cutaneous lesions. In all
patients,
the addition of methotrexate allowed reduction or discontinuation of other
therapies
such as prednisone. All patients tolerated the methotrexate with minimal
toxicity.
CONCLUSIONS: Low-dose oral methotrexate administered weekly is effective in
treatment
of the cutaneous manifestations of dermatomyositis and frequently enables a
reduction
or discontinuation of corticosteroid therapy.
=====================================================================
14.) Serum concentrations of carboxyterminal propeptide of type 1 procollogen
and tissue inhibitor of metalloproteinase 1 in patients with dermatomyositis.
=====================================================================
Author
Kubo M; Kikuchi K; Fujimoto M; Tamaki K
Address
Department of Dermatology, Faculty of Medicine, University of Tokyo, Japan.
kubo-der@h.u-tokyo.ac.jp
Source
Arch Dermatol Res, 290(5):253-7 1998 May
Abstract
Serum levels of carboxyterminal propeptide of type I procollagen (PICP) and
aminoterminal propeptide of type I procollagen (PINP) have been used as
indices of collagen synthesis in patients with various fibrotic diseases
during the active stages. One of the suggested contributory factors to the
development of tissue fibrosis is a decrease in collagenase activity, which
may be related to levels of serum tissue inhibitors of metalloproteinases-1
(TIMP-1). In this study, the serum levels of PICP and PINP in 20 patients
with dermatomyositis and 29 control subjects and of TIMP-1 in 29 patients
with dermatomyositis and 29 control subjects were measured using an
enzyme-linked immunosorbent assay (ELISA) or a radioimmunoassay (RIA). We
found that the mean PICP level in patients with dermatomyositis was
significantly higher than that in normal controls (mean +/- SD 326+/-76
ng/ml vs 135+/-88 ng/ml; P < 0.001). In 60% of dermatomyositis patients,
the serum PICP level was elevated (more than 311 ng/ml, i.e. 2 x SD above
the mean control value). Elevated serum PICP levels were correlated with
the incidence of elevated serum creatine kinase levels in patients with
dermatomyositis. The serum concentration of PINP was not elevated in
comparison with that of the normal control subjects (mean +/- SD 36+/-30
ng/ml vs 63+/-34 ng/ ml). The mean TIMP-1 level in the patients with
dermatomyositis was also significantly higher than in the normal control
subjects (mean +/- SD 438+/-328 ng/ml vs 163+/-63 ng/ml; P < 0.001). In 59%
of dermatomyositis patients, the mean serum TIMP-1 level was elevated (more
than 289 ng/ml, i.e. 2 x SD above the mean control value). Serum PICP and
TIMP-1 levels might be useful for detecting disease activity or severity in
dermatomyositis.
=====================================================================
15.) Dermatomyositis: a dermatology-based case series.
=====================================================================
Author
Dawkins MA; Jorizzo JL; Walker FO; Albertson D; Sinal SH; Hinds A
Address
Department of Dermatology, Bowman Gray School of Medicine of Wake Forest
University, Winston-Salem, North Carolina 27157-1071, USA.
Source
J Am Acad Dermatol, 38(3):397-404 1998 Mar
Abstract
BACKGROUND: Dermatomyositis is associated with significant morbidity and
occasional mortality. Currently there is no consensus on treatment for
patients with dermatomyositis. OBJECTIVE: Our purpose was to review the
clinical features and response to therapy of patients with dermatomyositis
and compare these data with previous series of patients with
dermatomyositis/polymyositis. METHODS: Clinical characteristics of 65
patients seen during a 10-year period were reviewed retrospectively.
Twenty-one of these patients were enrolled in a prospective, uncontrolled
study of treatment with high-dose prednisone followed by slow tapering.
RESULTS: Clinical features were similar to those previously described;
however, muscle strength at diagnosis was on average greater in patients in
this series than in patients previously reported. Malignancy was present in
5 of 43 adult patients (12%), but was not found in patients with juvenile
dermatomyositis. Another connective tissue disease was present in 19% of
patients. Twelve patients had dermatomyositis sine myositis. Eighteen of 21
patients (85%) in the prednisone study group had resolution of myositis.
CONCLUSION: Patients with dermatomyositis in this series had less active
myositis at presentation, but were otherwise similar to patients with
dermatomyositis/polymyositis previously reported. Treatment with high-dose
daily prednisone followed by slow tapering was effective.
=====================================================================
16.) Microvascular changes in skeletal muscle in idiopathic inflammatory
myopathy.
=====================================================================
Author
Estruch R; Grau JM; Fern´andez-Sol´a J; Casademont J; Monforte R;
Urbano-M´arquez A
Address
Department of Internal Medicine, Hospital Cl´inic, University of Barcelona,
Spain.
Source
Hum Pathol, 23(8):888-95 1992 Aug
Abstract
Open deltoid muscle biopsy specimens from patients with idiopathic adult
dermatomyositis, paraneoplastic dermatomyositis, childhood dermatomyositis,
and idiopathic polymyositis, and from control patients were studied.
Qualitative and morphometric capillary analysis by phase and electron
microscopy was carried out. In the morphologic analysis the most striking
difference was the presence of capillary damage and a higher capillary
depletion in dermatomyositis as well as a higher capillary density in
polymyositis. By electron microscopy, capillaries from patients with
dermatomyositis showed mainly microtubuloreticular structures, loss of
endothelial plasma membranes, and the appearance of abnormal cytoplasmic
organelles. In contrast, capillaries from patients with polymyositis
exhibited only minimal changes. By morphometric analysis, muscle
capillaries in dermatomyositis had a significantly higher mean endothelial
thickness than those in polymyositis. Finally, a significant topographic
association between capillary damage and muscle fiber changes was observed
only in patients with dermatomyositis. On the other hand, paraneoplastic
dermatomyositis showed fewer structural and morphometric capillary changes
than the other forms of dermatomyositis. We conclude that dermatomyositis
is characterized by microvascular alterations that are absent in
polymyositis. The topographic proximity of capillary changes to muscle
fiber injury suggests that capillary damage may play a role in the
pathogenesis of the muscle lesions observed in patients with dermatomyositis.
=====================================================================
17.) Risk of cancer in patients with dermatomyositis or polymyositis. A
population-based study.
=====================================================================
Author
Sigurgeirsson B; Lindel¨of B; Edhag O; Allander E
Address
Department of Dermatology, Karolinska Hospital, Stockholm, Sweden.
Source
N Engl J Med, 326(6):363-7 1992 Feb 6
Abstract
BACKGROUND. An association between polymyositis and cancer was first
proposed in 1916, but the existence of the association has been disputed.
An association between dermatomyositis and cancer is better accepted, but
its magnitude is not known. METHODS. We undertook a study to provide
accurate estimates of the risk of cancer in patients with dermatomyositis
or polymyositis. We studied the incidence of cancer and the rate of
mortality from cancer in a population-based cohort of 788 patients with
dermatomyositis or polymyositis in Sweden from 1963 through 1983. The
results were compared with those for the general population. RESULTS. Among
the 396 patients with polymyositis, 42 cancers were diagnosed at the same
time or after polymyositis was diagnosed in 37 patients (9 percent). The
relative risk of cancer was 1.8 (95 percent confidence interval, 1.1 to
2.7) in the male patients and 1.7 (95 percent confidence interval, 1.0 to
2.5) in the female patients. Eighty-four males and 85 females died, and in
24 of these cases (14 percent) cancer was the principal cause of death. The
mortality ratio (the rate of mortality from cancer in these patients as
compared with that in the general population) was 0.90 (95 percent
confidence interval, 0.6 to 1.4). Among the 392 patients with
dermatomyositis, 61 cancers were diagnosed at the same time or after
dermatomyositis was diagnosed in 59 patients (15 percent). The relative
risk of cancer was 2.4 (95 percent confidence interval, 1.6 to 3.6) in the
male patients and 3.4 (95 percent confidence interval, 2.4 to 4.7) in the
female patients. Fifty-seven males and 110 females died, and in 67 of these
cases (40 percent) cancer was the principal cause of death (mortality
ratio, 3.8; 95 percent confidence interval, 2.9 to 4.8). CONCLUSIONS. The
risk of cancer is increased in patients with polymyositis or
dermatomyositis. In patients with dermatomyositis there is also a higher
rate of mortality from cancer.
=====================================================================
18.) Ovarian malignancy in patients with dermatomyositis and polymyositis: a
retrospective analysis of fourteen cases.
=====================================================================
Author
Davis MD; Ahmed I
Address
Department of Dermatology, Mayo Clinic, Rochester, Minnesota, USA.
Source
J Am Acad Dermatol, 37(5 Pt 1):730-3 1997 Nov
Abstract
BACKGROUND: Dermatomyositis and polymyositis can be associated with an
underlying malignancy. OBJECTIVE: Our purpose was to describe a group of
patients with dermatomyositis and polymyositis in whom ovarian carcinoma
was diagnosed and to evaluate further the characteristics of the
association between these diseases. METHODS: A cross-sectional
retrospective review identified 14 patients in a 45-year period (1950 to
1995) with dermatomyositis and polymyositis in whom an underlying ovarian
malignancy was diagnosed. Clinical and laboratory data of these patients
were reviewed, and immunofluorescent and hematoxylin-eosin-stained skin
sections were examined. RESULTS: The mean age at diagnosis of
dermatomyositis and polymyositis was 59 years. Of the 14 patients, 11 had
dermatomyositis, two had polymyositis, and one had an overlap disease. The
12 patients with dermatomyositis had distinctive cutaneous lesions. In
eight, cutaneous biopsy specimens demonstrated variable histologic changes.
Thirteen patients had signs of proximal muscle weakness, and myopathy was
further demonstrated by electromyographic testing in 10. Creatine kinase
levels were increased in 6 of the 10 patients tested. Dysphagia was present
in five patients, but esophageal dysmotility was not demonstrated in three.
Dermatomyositis and polymyositis preceded the ovarian malignancy in nine
patients, were concomitant with it in four, and succeeded the ovarian
disease in one. CONCLUSION: Physical examination and imaging techniques
failed to detect early ovarian cancer in our patients with dermatomyositis
and polymyositis. When detected (usually by abdominopelvic examination, in
two by computed tomographic examination), it was advanced, and survival was
poor.
=====================================================================
19.) Breast cancer and second primary ovarian cancer in dermatomyositis.
=====================================================================
Author
Voravud N; Dimopoulos M; Hortobagyi G; Ross M; Theriault R
Address
Department of Medical Oncology, University of Texas M. D. Anderson Cancer
Center, Houston 77030.
Source
Gynecol Oncol, 43(3):286-90 1991 Dec
Abstract
We report six female patients with breast cancer who developed
dermatomyositis and compare our data with those from other reports. The
development of dermatomyositis in two patients led to the discovery of a
second primary ovarian carcinoma, whereas the development of
dermatomyositis in another two patients led to the discovery of recurrent
breast cancer. In three patients the diagnosis of dermatomyositis preceded
the diagnosis of breast cancer, while the rest developed dermatomyositis
after the diagnosis of breast cancer. A parallel clinical course of
dermatomyositis and breast cancer was seen in only one patient. Coexisting
dermatomyositis and breast cancer is a rare phenomenon, and dermatomyositis
that develops during the course of breast cancer may indicate the
occurrence of a second primary malignancy or recurrent breast cancer. The
onset of dermatomyositis may precede, coincide with, or follow the
diagnosis of breast cancer. The clinical course of dermatomyositis
sometimes, but not always, parallels the course of breast cancer. There are
no specific clinical or laboratory markers to distinguish patients with
dermatomyositis who have malignancy from those without cancer.
=====================================================================
20.) [Vascular manifestations of dermatomyositis and polymyositis. Clinical,
capillaroscopic and histological aspects]
=====================================================================
Author
Leteurtre E; Hachulla E; Janin A; Hatron PY; Brouillard M; Devulder B
Address
Service de m´edecine interne, CHRU, h^opital Claude-Huriez, Lille.
Source
Rev Med Interne, 15(12):800-7 1994
Abstract
Polymyositis is characterized by a T-cell-mediated and MHC-I-restricted
cytotoxic process, whereas dermatomyositis is a primitively vascular
disease with microangiopathy mediated by the complement C5b-9 membranolytic
attack complex. We have tried to estimate the frequency of vascular
abnormalities in polymyositis as defined by Bohan and Peter. We have
retrospectively studied 17 patients with dermatomyositis and 15 patients
with polymyositis. Vascular abnormalities have been defined by clinical,
capillaroscopic and histologic (muscle biopsy and minor salivary glands
biopsy) features. After clinical features, 5/17 dermatomyositis had a
Raynaud's phenomenon, against 6/15 polymyositis. Digital necrosis has been
observed for 2/17 dermatomyositis and 2/15 polymyositis. In capillaroscopy,
14/17 dermatomyositis had a microangiopathy with or no enlarged capillary
loops, against 7/15 polymyositis. None of these polymyositis had enlarged
capillary loops. The muscle biopsy showed a predominantly perivascular or
perimysial inflammatory infiltrate (vascular process) for 10/16
dermatomyositis against 4/13 polymyositis; a perifascicular atrophy for
3/16 dermatomyositis against 2/13 polymyositis. The histological study of
minor salivary glands, showed vascular lesions for 2/11 dermatomyositis and
for 1/8 polymyositis. Finally, Bohan and Peter's classification is now
inadequate to distinguish between dermatomyositis and polymyositis. Indeed,
some dermatomyositis sine dermatitis, may exist and be recognized by their
vascular features. To distinguish between dermatomyositis and polymyositis
is important, to evaluate the risk of cancer which is more frequent in
dermatomyositis.
=====================================================================
21.) Long-term prognosis of 69 patients with dermatomyositis or polymyositis.
=====================================================================
Author
Maugars YM; Berthelot JM; Abbas AA; Mussini JM; Nguyen JM; Prost AM
Address
Department of Rheumatology, Nantes University Hospital Center, France.
Source
Clin Exp Rheumatol, 14(3):263-74 1996 May-Jun
Abstract
OBJECTIVES: To assess the long-term prognosis of dermatomyositis and pol
myositis. METHODS: 69 patients with dermatomyositis or polymyositis were
selected according to the diagnostic criteria of Bohan and Peter and were
followed up for a minimum of 6.3 years (for surviving patients) (mean 11.6
years). Clinical and biological features, and pulmonary and muscle
parameters were considered as prognostic factors for death. Functional
disability was assessed using a 4-stage grading system. RESULTS: 30 deaths
(43.5%) occurred mainly due to cardiovascular (8), pulmonary (8),
carcinomatous (5) and iatrogenic complications (5). Survival rates were
82.6% at 1 year, 73.9% at 2.66, 7% at 5 and 55.4% at 9. Significant
prognostic factors for death (Cox model with time-dependent covariates)
were old age (p < 0.0001), dysphonia (p < 0.001), pulmonary interstitial
fibrosis (p < 0.02), absence of dysphagia (p < 0.02) and asthenia-anorexia
(p < 0.05). Dermatomyositis and polymyositis subgroups had slightly
different significant prognostic factors for death: old age, cancer,
pulmonary interstitial fibrosis and asthenia-anorexia for dermatomyositis;
old age, failure to improve muscle strength in response to treatment after
one month, and the absence of myalgia as presenting symptom for
polymyositis. At the end of the follow-up, 33/39 surviving patients (84.6%)
had no or insignificant muscular disability, whereas 3 children were
bedridden due to generalized calcinosis. CONCLUSIONS: High mortality
occurred in the first year, and the survival rate decreased continually up
to 9 years. The main prognostic factor for death is old age, but
dermatomyositis and polymyositis must be considered separately. General
features (pulmonary fibrosis, cancer, asthenia-anorexia) are involved in
dermatomyositis, whereas muscular symptoms are the most significant in
polymyositis. The long-term functional prognosis was fairly good, except
for generalized calcinosis, which tended to occur in childhood
dermatomyositis.
=====================================================================
22.) Dermatomyositis and malignancy.
=====================================================================
Author
Bernard P; Bonnetblanc JM
Address
Department of Dermatology, Dupuytren University Hospital, Limoges, France.
Source
J Invest Dermatol, 100(1):128S-132S 1993 Jan
Abstract
During the past 12 years, many studies applying strict diagnostic criteria
have been published that have attempted to settle the controversy about the
reality of the association between dermatomyositis and malignancy. Although
retrospective, recent studies have shown an increased incidence of
malignancy among patients with dermatomyositis when compared with controls
without myositis. In contrast, an increased frequency of malignancy in
dermatomyositis as compared to polymyositis still has to be demonstrated.
In most cases, malignant disease precedes or occurs concurrently with
dermatomyositis and is discovered on the basis of clinical signs, symptoms,
and routine screening laboratory tests. The types of neoplasms found in
association with dermatomyositis parallel those observed in the general
population. A possible link between dermatomyositis and an underlying
malignancy remains largely hypothetical at a biologic level, although
cellular immunity abnormalities may provide a direction for future
investigations. Prospective epidemiologic studies using the case-control
methods and cohort analysis remain necessary 1) to rigorously demonstrate
the reality and to study the nature of the association between
dermatomyositis and malignancy, and 2) to clarify the optimal screening
strategies for malignant neoplasms in patients with dermatomyositis.
=====================================================================
23.) Dermatomyositis. Disease associations and an evaluation of screening
investigations for malignancy.
=====================================================================
Author
Cox NH; Lawrence CM; Langtry JA; Ive FA
Address
Department of Dermatology, Royal Victoria Infirmary, Newcastle-upon-Tyne,
England.
Source
Arch Dermatol, 126(1):61-5 1990 Jan
Abstract
Fifty-three adult patients (19 men, 34 women) with dermatomyositis were
studied. Two had dermatomyositis associated with benign disorders.
Twenty-three (43%) had a malignancy; the risk of malignancy increased with
age, but there was no sex difference. Seven malignancies were recurrences
and 9 were diagnosed during investigation of dermatomyositis; these 16 were
suspected clinically or from abnormal results of simple investigations.
Extensive screening tests did not increase the number of malignancies
diagnosed. In 7 patients, a diagnosis of malignancy was made more than 9
months after onset of dermatomyositis, although a relationship between
malignancy and dermatomyositis was uncertain in two cases; the diagnosis of
gynecological malignancy was missed in 2 patients despite appropriate
investigations, 1 patient had poorly controlled dermatomyositis, and in 2
patients late diagnosis of malignancy was due to failure to reinvestigate
relapse of previously stable dermatomyositis.
=====================================================================
24.) [Dermatomyositis complicated by sarcoidosis]
=====================================================================
Author
Takano Y; Oida K; Kohri Y; Taguchi Y; Tomii K; Matsumura Y; Mino M; Gohma
I; Kobashi Y; Yuba Y
Address
Department of Respiratory Medicine and Pathology, Tenri Hospital.
Source
Nihon Kyobu Shikkan Gakkai Zasshi, 34(11):1255-9 1996 Nov
Abstract
We encountered a patient with dermatomyositis complicated by sarcoidosis. A
57-year-old woman was admitted to our hospital because of fever dry cough,
and myalgias. There were reticular shadows on her chest X-ray film.
Although the typical skin rash and myositis suggested the diagnosis of
dermatomyositis biopsy specimens from a salivary gland, muscle, and lung
revealed noncaseating granulomas as well. Uveitis was also noted. These
findings suggested the coexistence of sarcoidosis with dermatomyositis.
Examination of the lung-biopsy specimens showed interstitial pneumonia
compatible with dermatomyositis, except for the granuloma. The typical rash
of dermatomyositis and pathological findings of the lung specimen were
inconsistent with sarcoidosis. Therefore we concluded that this patient had
both dermatomyositis and sarcoidosis. This case sheds new light on the
importance of pathological examinations.
=====================================================================
25.) [Prognostic factors and predictive signs of malignancy in adult
dermatomyositis]
=====================================================================
Author
Gallais V; Crickx B; Belaich S
Address
Service de Dermatologie, H^opital Bichat, Paris.
Source
Ann Dermatol Venereol, 123(11):722-6 1996
Abstract
INTRODUCTION: Prognosis in dermatomyositis is severe, partly due to the
development of cancer. The aim of this study was to identify factors
predicting cancer development and assess factors predicting reduced
survival rate. PATIENTS AND METHODS: A retrospective analysis of 32 cases
of dermatomyositis diagnosed on the basis of the Bohan and Peter criteria
was performed. Diagnosis was certain in 7 cases, probable in 13 and
possible in 5. There were thus 7 cases of pure cutaneous dermatomyositis.
Clinical and laboratory data were compared between patients with and
without cancer and between deceased and surviving patients. RESULTS:
Overall mortality was 37.5% at 4 years, confirming the gravity of
dermatomyositis. Malignancy developed in 9 patients (28.1%) leading to
death in all cases, within 18 months in 8. Amyopathic dermatomyositis was
observed in 2 of these patients. Necrotic skin ulcerations (p < 0.01) and
pruritis (p < 0.05) were significant predictive factors for the development
of cancer. Poor prognosis factors were malignancy (p < 0.001), necrotic
skin ulcerations (p < 0.01), and pruritis (p < 0.05). CONCLUSION: Prognosis
is poor in certain sub-groups of patients with dermatomyositis. Such
patients can be identified on the basis of skin lesions, notable necrotic
ulcerations.
=====================================================================
26.) Scalp involvement in dermatomyositis. Often overlooked or misdiagnosed.
=====================================================================
Author
Kasteler JS; Callen JP
Address
Division of Dermatology, University of Louisville, School of Medicine, KY.
Source
JAMA, 272(24):1939-41 1994 Dec 28
Abstract
OBJECTIVE--To characterize scalp involvement in patients with
dermatomyositis. DESIGN--Case series. PATIENTS--All patients with
dermatomyositis seen in our office between 1988 and mid 1993. Patient
inclusion in this study required fulfillment of three or more of Bohan and
Peter's criteria for dermatomyositis. RESULTS--Of 17 patients with the
diagnosis of dermatomyositis, scalp involvement was present in 14. Five of
the 14 patients with scalp involvement were diagnosed as having scalp
psoriasis or seborrheic dermatitis before progression of their disease or
tissue examination revealed the diagnosis of dermatomyositis. In all
patients, the scalp involvement was manifested as atrophic, erythematous,
scaly plaques. In addition, alopecia was noted in six of the 14 patients.
Treatment of cutaneous involvement included sun avoidance, topical
corticosteroids and/or antimalarials, and/or methotrexate.
CONCLUSIONS--Recognition of this process is important because scalp
involvement is often overlooked, may be misdiagnosed initially, and can be
the presenting complaint in some patients with dermatomyositis.
=====================================================================
27.) Cutaneous changes of dermatomyositis in patients with normal muscle
enzymes: dermatomyositis sine myositis?
=====================================================================
Author
Stonecipher MR; Jorizzo JL; White WL; Walker FO; Prichard E
Address
Department of Dermatology, Bowman Gray School of Medicine, Wake Forest
University, Winston-Salem, NC 27157.
Source
J Am Acad Dermatol, 28(6):951-6 1993 Jun
Abstract
BACKGROUND: Dermatomyositis sine myositis may have various connotations.
Controversy exists as to nomenclature, degree of evaluation required,
therapy, and course (e.g., does true dermatomyositis of the skin only
exist?). OBJECTIVE: The purpose of this study was to assess prospectively
patients with the clinicopathologic features of dermatomyositis and normal
muscle enzyme serum levels to determine their course in terms of the onset
of muscle disease. METHODS: Thirteen patients were studied by complete
history and clinical examination, laboratory studies, electromyography, and
skin and muscle biopsy. They were observed for 1 to 6 years. RESULTS:
Patients were classifiable into three groups: (1) cutaneous changes only,
(2) cutaneous changes only at baseline with subsequent development of
myositis, and (3) cutaneous changes with normal muscle enzyme serum levels
at baseline but with myositis demonstrated by electromyography and/or
muscle biopsy specimens. CONCLUSION: Significantly different prognostic and
therapeutic implications are present in patients with dermatomyositis with
normal muscle enzyme serum levels depending on the results of
electromyography, muscle biopsy, and clinical observation.
=====================================================================
28.) Association between bovine collagen dermal implants and a dermatomyositis
or a polymyositis-like syndrome [see comments]
=====================================================================
Author
Cukier J; Beauchamp RA; Spindler JS; Spindler S; Lorenzo C; Trentham DE
Address
Baylor College of Medicine, Department of Plastic Surgery, Houston, TX 77030.
Source
Ann Intern Med, 118(12):920-8 1993 Jun 15
Abstract
OBJECTIVE: To determine whether an excess incidence of dermatomyositis or
polymyositis or both exist in patients treated with injectable bovine
collagen implants and to characterize the clinical picture. DESIGN:
Historical cohort study (July 1980 through June 1988). PATIENTS: Patients
were identified from personal experience or adverse reaction reports
received by the manufacturer. SETTING: An 8-year period in the United
States during which approximately 345,000 patients received implants.
RESULTS: Eight patients with dermatomyositis and an additional patient with
polymyositis were identified from approximately 345,000 patients receiving
injectable bovine collagen implants from July 1980 through June 1988. The
nine patients with dermatomyositis or polymyositis were diagnosed an
average of 6.4 months (range, 0.7 to 24.9 months) after collagen implant or
skin test exposure or both. Eight of the nine patients had a delayed-type
hypersensitivity response at the test or treatment sites or both, and five
of six patients tested were found to have increased serum antibodies to
collagen. Compared with the general population, the incidence of
dermatomyositis or polymyositis among collagen-treated patients was
statistically increased (standardized incidence ratio, 5.05; 95% CI, 2.31
to 9.59; P < 0.0001). A similar analysis of the eight dermatomyositis case
patients produced a standardized incidence ratio of 18.8 (CI, 8.1 to 37.0;
P < 0.0001). Using a Monte Carlo simulation, an interval of 6.4 months or
less from exposure to onset of disease was found to be an extremely rare
event, occurring less than 72 times per one million simulation trials (CI,
57 to 91). CONCLUSIONS: Because these data suggest that an immunologic
response to bovine type I or type III collagen or both caused this
dermatomyositis or polymyositis-like syndrome, the risks versus benefits
for the cosmetic use of collagen implants should be reassessed.
=====================================================================
29.) MR imaging in amyopathic dermatomyositis.
=====================================================================
Author
Lam WW; Chan H; Chan YL; Fung JW; So NM; Metreweli C
Address
Department of Diagnostic Radiology and Organ Imaging, Chinese University of
Hong Kong, Shatin, Hong Kong.
Source
Acta Radiol, 40(1):69-72 1999 Jan
Abstract
PURPOSE: Amyopathic dermatomyositis is a distinct clinical entity with
cutaneous involvement but no myopathy. We conducted a prospective study to
investigate the role of MR imaging in these patients. MATERIAL AND METHODS:
Out of 40 Chinese patients presenting with dermatomyositis, based on
clinical assessment and normal serum muscle enzymes 10 were diagnosed as
having amyopathic dermatomyositis. These 10 patients underwent MR imaging
for evaluation of any subclinical muscle involvement. RESULTS: Three
patients demonstrated abnormal signal intensity in muscles on both T2- and
fat suppression sequences. Thus, one-third of patients with dermatomyositis
and clinically normal muscles may have detectable muscle inflammation on MR
images, indicating that MR has a potential role for locating the relevant
biopsy site and for longitudinal follow up. Six of the 10 patients had
malignant disease diagnosed before or after diagnosis of the cutaneous
manifestation. Nasopharyngeal carcinoma was the most common malignant
disease in this group of patients. CONCLUSION: MR imaging is recommended
for demonstrating subclinical muscle involvement in patients with the
clinical diagnosis of amyopathic dermatomyositis. We also recommend
screening for malignancy, particularly nasopharyngeal carcinoma, in
Southern Chinese patients with dermatomyositis.
=====================================================================
30.) Case Report: amyopathic dermatomyositis associated with transformed
malignant lymphoma.
=====================================================================
Author
Osman Y; Narita M; Kishi K; Fujiwara H; Fukuda T; Koike T; Shibata A
Address
First Department of Internal Medicine, Niigata University School of
Medicine, Japan.
Source
Am J Med Sci, 311(5):240-2 1996 May
Abstract
Amyopathic dermatomyositis is a disease of unknown origin characterized by
the specific skin lesions of dermatomyositis but without clinical or
laboratory evidence of myopathy. During the past 15 years, a great
controversy between the different reports concerning a possible association
of dermatomyositis with malignancy has been noted. In this report, the
authors describe a patient with amyopathic dermatomyositis who presented
first with a benign hyperplasia of the lymph node, which finally
transformed into frank malignant lymphoma. In addition to follow-up care,
screening tests to search for occult malignancy in patients with amyopathic
dermatomyositis (or dermatomyositis) are recommended.
=====================================================================
31.) Malignancy in adult dermatomyositis.
=====================================================================
Author
Leow YH; Goh CL
Address
National Skin Centre, Singapore.
Source
Int J Dermatol, 36(12):904-7 1997 Dec
Abstract
BACKGROUND: Dermatomyositis has been reported to be associated with
malignancies in 15%-34% of patients in Western countries, but in as many as
two-thirds of patients in Singapore. The aim of this study was to determine
whether a diagnostic measure could be helpful in the diagnosis of a
malignancy in patients with dermatomyositis. METHODS: This is a
retrospective study on 38 adult patients with dermatomyositis that were
seen over a 6-year period from 1989 to 1994. RESULTS: All the patients
presented with cutaneous features that suggest the clinical diagnosis of
dermatomyositis; however, not all cases show all the key features of the
disease. Of the studied patients, 86.8% were noted to have photosensitivity
as a key cutaneous presentation. Thirty (78.9%) of our patients were above
the age of 40 years, and 12 (31.6%) of these were found to have an
associated malignancy. Nasopharyngeal carcinoma was the most commonly
associated malignancy (38.4%) in our study population. CONCLUSIONS: In our
study population, otorhinolaryngologic screening is an essential
investigation for the evaluation of dermatomyositis in association with
malignancy.
=====================================================================
32.) Treacher-Collins syndrome and co-existing dermatomyositis.
=====================================================================
Author
Larenas-Linnemann DE; Berr´on-Perez R; Ortega-Martell JA; Onuma-Takane E;
Huicochea-Grobet Z
Address
Instituto Nacional de Pediatria, Mexico City, Mexico.
Source
Ann Allergy Asthma Immunol, 80(1):50-4 1998 Jan
Abstract
BACKGROUND: Treacher-Collins syndrome, an autosomal dominantly inherited
malformation of structures derived from the first and second branchial
arch, has an incidence of 1:10,000 newborns. The prevalence of
dermatomyositis at less than 24 years of age has been estimated at 1 per
100,000. The occurrence of both Treacher-Collins syndrome and
dermatomyositis combined in the same patient should occur once in every
1,000,000,000 subjects. METHODS: We report a patient with Treacher-Collins
syndrome who developed dermatomyositis at the age of 5 years. RESULTS: No
other patient with both Treacher-Collins syndrome and an autoimmune disease
has been reported. The thymus originates from the third branchial pouch and
is unaffected by the syndrome. In Treacher-Collins syndrome the affected
gene has been mapped to the fifth chromosome, while dermatomyositis is
related to HLA B8 and DR3, coded on the sixth chromosome. No immunologic
alteration has been described in patients with Treacher-Collins syndrome.
CONCLUSION: This is the first report of a patient with Treacher-Collins
syndrome and dermatomyositis. There is no genetic or physiopathologic
explanation for the concurrence of both conditions.
=====================================================================
33.) [A clinical analysis of cutaneous type dermatomyositis]
=====================================================================
Author
Yu B
Address
PUMC Hospital, CAMS, Beijing.
Source
Chung Kuo I Hsueh Ko Hsueh Yuan Hsueh Pao, 16(5):394-6 1994 Oct
Abstract
This paper reports nine patients with the classic cutaneous findings of
dermatomyositis who did not develop clinical or laboratory evidence of
muscle disease for at least 2 years after onset of their skin
manifestations. Such patients represent 3.5% of our total experience with
dermatomyositis patients during a 12 years period. None of the patients had
evidence of malignancy. Each of five patients treated with oral prednisone
for their cutaneous lesion or mild myositis after onset of their skin
manifestations 3-12 years and had marked improvement. The author emphasizes
that the cutaneous manifestations of dermatomyositis are pathognomonic of
this disease and the term of this disease proposes cutaneous type
dermatomyositis better than amyopathic dermatomyositis.
=====================================================================
34.) Dermatomyositis with a pityriasis rubra pilaris-like eruption: a
little-known distinctive cutaneous manifestation of dermatomyositis.
=====================================================================
Author
Requena L; Grilli R; Soriano L; Escalonilla P; Fari~na C; Mart´in L
Address
Department of Dermatology, Fundaci´on Jim´enez D´iaz, Universidad
Aut´onoma, Madrid, Spain.
Source
Br J Dermatol, 136(5):768-71 1997 May
Abstract
A pityriasis rubra pilaris-like eruption has been described in patients
with dermatomyositis. These patients showed generalized follicular
hyperkeratosis and diffuse thickening of the palms and soles.
Histopathological findings consisted of keratotic plugging of the
follicular infundibulum and features of erector pili myositis. We report on
an 18-year-old woman with dermatomyositis. The diagnosis was established by
characteristic enzymatic alterations, electromyographic pattern of myositis
and the findings in a muscle biopsy, although the patient had no evidence
of muscular weakness during a follow-up of 14 years. She developed an
erythematosus and squamous eruption associated with diffuse palmoplantar
keratoderma. Histopathological features consisted of a papillomatous
epidermis with spicules of compact eosinophilic hyperkeratosis over the
tips of papillae that were not related to hair follicles. Pityriasis rubra
pilaris-like eruption seems to be a characteristic although uncommon
cutaneous manifestation in dermatomyositis.
=====================================================================
35.) HCV and dermatomyositis: report of 5 cases of dermatomyositis in patients
with HCV infection.
=====================================================================
Author
Fiore G; Giacovazzo F; Giacovazzo M
Address
VI Medical Clinic, University La Sapienza, Rome, Italy.
Source
Riv Eur Sci Med Farmacol, 18(5-6):197-201 1996 Sep-Dec
Abstract
The authors report five cases of dermatomyositis in which positivity for
hepatitis C virus was ascertained. A virus-related (ECHO virus, Coxsackie)
dermatomyositis is known, but a hepatitis virus C-related dermatomyositis
was reported only in one case in a Letter to the Editor of Journal of
Rheumatology (1994).
=====================================================================
36.) [Dermatomyositis and cancer. A retrospective study]
=====================================================================
Author
Selvaag E; Thune P; Austad J
Address
Hudavdelingen Ullev al sykehus, Oslo.
Source
Tidsskr Nor Laegeforen, 114(20):2378-80 1994 Aug 30
Abstract
In a retrospective study, the files of 19 patients with dermatomyositis
examined at our departments from 1970 to 1993 were reviewed. The parameters
studied were age, sex, muscle enzyme values, muscle biopsies,
electromyographical findings and interval from onset of dermatomyositis
until first visit to the department. Out of 19 patients with
dermatomyositis, 18 were adults and in nine of these the condition was
associated with cancer (three out of three men, six out of 15 women).
Electromyographical findings were pathological in 17 investigated patients
and myositis was indicated in 13 out of 15 biopsies. Muscle enzyme values
were elevated in seven out of nine patients with cancer and in three out of
nine without. Out of five patients with dysphagia, four patients had
cancer. The risk of cancer is increased in patients with dermatomyositis.
Factors indicating a poor prognosis regarding the association between
dermatomyositis and cancer in our study were old age, male sex and dysphagia.
=====================================================================
37.) Amyopathic dermatomyositis (dermatomyositis sin´e myositis). Presentation
of six new cases and review of the literature [see comments]
=====================================================================
Author
Euwer RL; Sontheimer RD
Address
Department of Dermatology, University of Texas Southwestern Medical Center,
Dallas, TX 75235.
Source
J Am Acad Dermatol, 24(6 Pt 1):959-66 1991 Jun
Abstract
We report six patients with the classic cutaneous findings of
dermatomyositis who did not develop clinical or laboratory evidence of
muscle disease for at least 2 years after onset of their skin
manifestations. Such patients represent 11% of our total experience with
dermatomyositis patients during a 15 year period. All six patients had
Gottron's paules, periungual erythema/telangiectasia, and violaceous
discoloration of the face, neck, upper chest, and back at some time during
the course of their disease. In addition, all complained of pruritus and
photosensitivity. None of the patients had evidence of malignancy. Each of
five patients treated with oral corticosteroids for their cutaneous disease
had marked improvement and did not develop myositis. These cases further
emphasize that the cutaneous manifestations of dermatomyositis are
pathognomonic of this disease and challenge the commonly held notion that
muscle disease always develops within 2 years of onset of skin disease.
======================================================================
38.) Epstein-Barr virus strain type and latent membrane protein 1 gene
deletions in
lymphomas in patients with rheumatic diseases.
======================================================================
Natkunam Y; Elenitoba-Johnson KS; Kingma DW; Kamel OW
Stanford University School of Medicine, California, USA.
Arthritis Rheum (UNITED STATES) Jun 1997 40 (6) p1152-6 ISSN: 0004-3591
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9708
Subfile: AIM; INDEX MEDICUS
OBJECTIVE: Recent studies have shown that immunomodulatory therapy for the
treatment of rheumatic diseases can be associated with the development of
Epstein-
Barr virus (EBV)-associated lymphoproliferative disorders. The present
study was
undertaken to determine the strain type of EBV in lymphoproliferative
disorders that
occur in patients with rheumatic disease and to investigate EBV latent
membrane
protein 1 (LMP-1) gene deletions that occur in these lymphoproliferative
disorders.
METHODS: Ten EBV-associated lymphoid neoplasms in patients with rheumatoid
arthritis
or dermatomyositis were analyzed by polymerase chain reaction to determine
EBV strain
type and to investigate for the presence of a previously characterized
30-basepair
deletion in the LMP-1 gene. RESULTS: The results indicated that
lymphoproliferative
disorders in these patients can harbor EBV strain type A or B, with a
predominance of
type A infection (80%). It was also shown that both wild-type and mutated
LMP-1
genes can be found in these neoplasms, with the deleted form of the LMP-1
gene
occurring in one-third of cases in this series. CONCLUSION: LMP-1 deletions
associated with certain aggressive lymphoid neoplasms are not required for
the
genesis of lymphoproliferative disorders in patients with rheumatic
disease. The
relative frequencies of type A and type B EBV strains in these
lymphoproliferative
disorders show similarities to the frequencies in patients with post-solid
organ
transplantation immunosuppression-associated lymphoproliferative disorders.
=====================================================================
DATA-MÉDICOS/DERMAGIC-EXPRESS No (55) 13/05/99 DR. JOSE LAPENTA R.
=====================================================================
Produced by Dr. José Lapenta R. Dermatologist
Venezuela
1.998-2.024
Producido por Dr. José Lapenta R. Dermatólogo Venezuela 1.998-2.0024
Tlf: 0414-2976087 - 04127766810












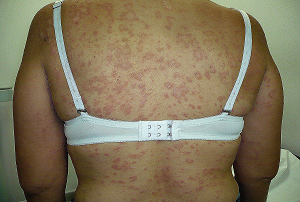




.png)