
PAPULOSIS LINFOMATOIDE, ACTUALIZACIÓN



Hola amigos de la red, DERMAGIC de nuevo con ustedes, el tema de hoy es LA PAPULOSIS LINFOMATOIDE (PL) enfermedad de la piel que bien está demostrado es precursora de malignidad (LINFOMA,). Su relación con la PITIRIASIS LIQUENOIDE AGUDA (PLEVA) y CRÓNICA (PLC) también está demostrada y bien documentada.
Considerada hoy día como una patología del tipo linfoproliferativo, con lesiones cutáneas, que son benignas, pero que histopatológicamente muestran rasgos de malignidad. El tratamiento es difícil.
HISTORIA:
La papulosis linfomatoide (PL) fue descrita por primera vez por Macaulay en 1968, quien la definió como un trastorno caracterizado por "una erupción continua de lesiones cutáneas con autoreparación, que parecen benignas clínicamente, pero que muestran rasgos histológicos malignos".
El científico propuso el nombre de PAPULOSIS LINFOMATOIDE (PL); transcurrieron casi 20 años hasta la década de los 80 cuando se identificó el marcador CD30 en las biopsias de lesiones, y posteriormente en 1997 fue cuando la OMS la reconoció como entidad nosológica con ese nombre.
CARACTERÍSTICAS CLÍNICAS:
La PAPULOSIS LINFOMATOIDE (PL) se presenta principalmente como pápulas o nódulos eritematosos, los cuales pueden ulcerarse, con diámetro variado desde milímetros a varios centímetros. Por lo general se presentan en el tronco y extremidades, pero pueden aparecer en cualquier parte del cuerpo, afecta mayormente adultos, pero también puede presentarse en adolescentes y niños.
Las lesiones son de tipo crónico y recurrentes, es decir evolucionan por brotes, y tardan en sanar unas 3 a 12 semanas espontáneamente, dejando cicatrices en el sitio de las lesiones las cuales pueden ser hiperpigmentadas o atróficas.
Los síntomas locales son leves: prurito y sistémicos no los hay. La evolución de los brotes es variada no presentándose un patrón específico.
DIAGNÓSTICO:
El diagnóstico definitivo de la PAPULOSIS LINFOMATOIDE (PL), ademas de las características clínicas ya descritas están basados en la biopsia cutánea la cual reporta:
Un Infiltrado dérmico compuesto principalmente por linfocitos T atípicos grandes con núcleos pleomórficos, redondeados y núcleos agrandados similares a las células de Reed-Sternberg (como en linfoma de Hodgkin).
Hay células con núcleos cerebriformes, similares a los observados en la MICOSIS FUNGOIDE (MF) o LINFOMA CUTANEO DE CELULAS T, y el infiltrado se observa TÍPICAMENTE en forma de CUÑA, con el vértice hacia abajo (dermis papilar).
Puede encontrarse también, edema de la dermis papilar, espongiosis, presencia de neutrofilos, eosinofilos y macrofagos.
Pero el DIAGNÓSTICO DEFINITIVO se hace mediante una prueba histoquímica para detectar las CÉLULAS CD30 POSITIVAS, en la biopsia.
El marcador CD30 es una glicoproteína que se expresa principalmente en linfocitos T y B activado, el cual es típico encontrarlo en enfermedades malignas, como el linfoma cutáneo de células T (micosis fungoides) y en las células tumorales de la mayoría de los linfomas.
DIAGNÓSTICO DIFERENCIAL:
Hay que recordar aquí, que el 10 a 20% de los casos de PAPULOSIS LINFOMATOIDE (PL), pueden evolucionar a MALIGNIDAD, tipo linfomas. Por otra parte la mayoría de los casos 90 a 80% suele remitir, es decir es considerada una afección crónica recurrente pero BENIGNA, con un pronóstico de SUPERVIVENCIA a los 10 años del 100%, excepto los casos que presentan evolución tórpida.
Los diagnósticos diferenciales son:
- PITIRIASIS LIQUENOIDE: AGUDA (PLEVA) o CRÓNICA (PLC).
- INFECCIONES CUTÁNEAS O SISTÉMICAS: IMPÉTIGO CONTAGIOSO, ENFERMEDAD DE LYME (BORRELIOSIS), HEPATITIS B y C, CITOMEGALOVIRUS (CMV), EPSTEIN BARR VIRUS (EBV), HIV.
- MICOSIS FUNGOIDE (MF): LINFOMA CUTANEO DE CELULAS T.
- LINFOMA LINFOCÍTICO T CUTÁNEO PRIMARIO: DE CÉLULAS PEQUEÑAS O MEDIANAS.
- ENFERMEDAD DE HODGKIN.
- LINFOMA ANAPLÁSICO CUTÁNEO DE CÉLULAS GIGANTES: (LACG-C).
- PSEUDOLINFOMAS.
- REACCIONES INFLAMATORIAS: PICADURAS DE INSECTOS, PSORIASIS, ERUPCIÓN POR DROGAS.
TRATAMIENTOS:
A.- TRATAMIENTOS CLÁSICOS:
Hasta hoy día, la papulosis linfomatoide (PL) se sigue tratando con las terapéuticas clásicas:
- Fototerapia y metotrexato: tratamiento de primera línea.
- Corticosteroides tópicos.
- Observación de la evolución de la enfermedad.
NUEVOS TRATAMIENTOS:
A. - NUEVOS RETINOIDES: BEXAROTENO: Cuya presentación es en forma oral y tópica, con el nombre comercial de TARGRETIN, aprobado para su uso en el Linfoma Cutáneo de Células T (CTCL), y es usado en la PAPULOSIS LINFOMATOIDE, en casos refractarios a la fototerapia y metotrexato, mostrando eficacia en la resolución de los brotes y disminución de las lesiones.
B.- INMUNOMODULADORES:
- INTERFERÓN ALFA: Para casos resistentes, puede usarse en combinación con fototerapia.
- IMIQUIMOD TÓPICO: empleado en lesiones localizadas., estimulando la respuesta inmune.
C.- RADIOTERAPIA LOCALIZADA: Utilizada en lesiones únicas persistentes, que no responden a otros tratamientos.
D.- PRODUCTOS BIOLÓGICOS:
- ANTICUERPOS MONOCLONALES: con efecto anti-CD30: BRENTUXIMAB VEDOTIN, utilizado en los linfomas cutáneos CD30+ (positivos). Se ha utilizado en la Papulosis Linfomatoide en casos excepcionales, las cuales presentan una evolución o transformación a linfomas.
- ONTAK: DAB(389)-IL-2: es una proteína de fusión terapéutica que combina la toxina diftérica (DAB-389) con la interleucina-2 (IL-2, bajo el nombre comercial de ONTAK (denileukin diftitox), utilizado fundamentalmente en Linfomas Cutaneos de celulas T. Utilizado experimentalmente en la Papulosis Linfomatoide (PL), casos refractarios.
E.- POLIQUIMIOTERAPIA SISTÉMICA: Utilizada en casos refractarios con evolución a linfoma. Los ciclos son de corta duración.
RESUMEN:
La PAPULOSIS LINFOMATOIDE (PL) es una enfermedad de difícil tratamiento, porque siendo considerada una enfermedad benigna en la mayoría de los casos, puede evolucionar en ciertos casos a MALIGNIDAD, tipo LINFOMA, por lo tanto el tratamiento debe elegirse de acuerdo a cada caso en particular.
El tratamiento de elección es el METOTREXATO, la FOTOTERAPIA, y los ESTEROIDES TÓPICOS, las otras terapias descritas se utilizan en casos refractarios y /o lesiones solitarias.
Saludos a todos !!!
Dr. José Lapenta R.,,,
EDITORIAL ENGLISH:
Hello friends of the network, DERMAGIC is back with you again. Today's topic is LYMPHOMATOID PAPULOSIS (LP), a skin disease that has been well established as a precursor to malignancy (LYMPHOMA). Its relationship with Pityriasis Lichenoides Acute (PLEVA) and Chronica (PLC) is also proven and well documented.
Today, it is considered a lymphoproliferative disease, with benign skin lesions that histopathologically show malignant features. Treatment is difficult.
HISTORY:
LYMPHOMATOID PAPULOSIS (LP) was first described by Macaulay in 1968, who defined it as a disorder characterized by "a continuous eruption of self-healing skin lesions that appear clinically benign but show malignant histological features."
The scientist proposed the name LYMPHOMATOID PAPULOSIS (LP); It took almost 20 years until the 1980s when the CD30 marker was identified in lesion biopsies, and later, in 1997, the WHO recognized it as a nosological entity with that name.
CLINICAL CHARACTERISTICS:
LYMPHOMATOID PAPULOSIS (LP) primarily present as erythematous papules or nodules, which may ulcerate, with diameters ranging from millimeters to several centimeters. They usually appear on the trunk and extremities, but can occur anywhere on the body. They primarily affect adults, but can also occur in adolescents and children.
The lesions are chronic and recurrent, that is, they develop in outbreaks and take 3 to 12 weeks to heal spontaneously, leaving scars at the lesion site, which may be hyperpigmented or atrophic.
Local symptoms are mild: pruritus, and there are no systemic symptoms. The course of outbreaks varies, with no specific pattern.
DIAGNOSIS:
The definitive diagnosis of LYMPHOMATOID PAPULOSIS (LP), in addition to the clinical features already described, is based on a skin biopsy, which reveals:
A dermal infiltrate composed primarily of large atypical T lymphocytes with pleomorphic, rounded nuclei and enlarged nuclei similar to Reed-Sternberg cells (as in Hodgkin lymphoma).
There are cells with cerebriform nuclei, similar to those seen in MYCOSIS FUNGOIDE (MF) or CUTANEOUS T-CELL LYMPHOMA, and the infiltrate is typically WEDGE-shaped, with the apex pointing downward (papillary dermis).
Edema of the papillary dermis, spongiosis, and the presence of neutrophils, eosinophils, and macrophages may also be present.
However, the DEFINITIVE DIAGNOSIS is made through a histochemical test to detect CD30+ positive cells in the biopsy.
The CD30 marker is a glycoprotein expressed primarily in activated T and B lymphocytes, and is typically found in malignant diseases, such as cutaneous T-cell lymphoma (mycosis fungoides) and in the tumor cells of most lymphomas.
DIFFERENTIAL DIAGNOSIS:
It should be noted here that 10 to 20% of cases of LYMPHOMATOID PAPULOSIS (LP) can progress to LYMPHOMA-LIKE malignancy. Furthermore, most cases (90 to 80%) usually regress, meaning it is considered a chronic, relapsing but benign condition, with a 10-year survival prognosis of 100%, except for cases that present a torpid progression.
Differential diagnoses include:
- PITYRIASIS LICHENOIDES: ACUTE (PLEVA) or CHRONIC (PLC).
- CUTANEOUS OR SYSTEMIC INFECTIONS: : CONTAGIOUS IMPETIGO, LYME DISEASE (BORRELIOSIS), HEPATITIS B and C, CYTOMEGALOVIRUS (CMV), EPSTEIN-BARR VIRUS (EBV), HIV.
- MYCOSIS FUNGOIDE (MF): CUTANEOUS T-CELL LYMPHOMA.
- PRIMARY CUTANEOUS T-LYMPHOCYTIC LYMPHOMA: small or medium-sized cells.
- HODGKIN'S DISEASE.
- CUTANEOUS ANAPLASTIC GIANT CELL LYMPHOMA: (C-ALCL).
- PSEUDOLYMPHOMAS.
- INFLAMMATORY REACTIONS: INSECT BITES, PSORIASIS, DRUG RASH.
TREATMENTS:
A.- CLASSIC TREATMENTS:
To this day, lymphomatoid papulosis (LP) is still treated with classic therapies:
- Phototherapy and methotrexate: first-line treatment.
- Topical corticosteroids.
- Monitoring disease progression.
NEW TREATMENTS:
A.- NEW RETINOIDS: BEXAROTENE: Available in oral and topical forms under the brand name TARGRETIN, it is approved for use in Cutaneous T-Cell Lymphoma (CTCL) and is used in LYMPHOMATOID PAPULOSIS in cases refractory to phototherapy and methotrexate, showing efficacy in resolving flares and reducing lesions.
B.- IMMUNOMODULATORS:
- INTERFERON ALPHA: For resistant cases, it can be used in combination with phototherapy.
- TOPICAL IMIQUIMOD: Used for localized lesions, stimulating the immune response.
C.- LOCALIZED RADIOTHERAPY: Used for persistent, single lesions that do not respond to other treatments.ents.
D.- BIOLOGICAL PRODUCTS:
- MONOCLONAL ANTIBODIES: with anti-CD30 effect: BRENTUXIMAB VEDOTIN, used in CD30+ (positive) cutaneous lymphomas. It has been used in lymphomatoid papulosis in exceptional cases, which present with progression or transformation into lymphomas.
- ONTAK: DAB(389)-IL-2: is a therapeutic fusion protein that combines diphtheria toxin (DAB-389) with interleukin-2 (IL-2, under the trade name ONTAK (denileukin diftitox). It is primarily used in cutaneous T-cell lymphomas. It has also been used experimentally in refractory cases of lymphomatoid papulosis (LP).
E.- SYSTEMIC POLYCHEMOTHERAPY: Used in refractory cases that progress to lymphoma. Cycles are short.
SUMMARY:
LYMPHOMATOID PAPULOSIS (LP) is a difficult-to-treat disease because, although it is considered benign in most cases, it can progress to lymphoma-like malignancy in certain cases. Therefore, treatment should be chosen based on each particular case.
The treatment of choice is METHOTREXATE, PHOTOTHERAPY and TOPICAL STEROIDS, the other therapies described are used in refractory cases and/or solitary lesions.
Greetings to all!!!
Dr. José Lapenta R.
===============================================================
REFERENCIAS BIBLIOGRÁFICAS / BIBLIOGRAPHICAL REFERENCES
================================================================
A.- First-Line Treatment in Lymphomatoid Papulosis: A Retrospective Multicentre Study (2018).
F.- Treatment of lymphomatoid papulosis with imiquimod 5% cream (2006).
G.- Lymphomatoid papulosis responding to topical methotrexate (2021).
H.- The efficacy of methotrexate for lymphomatoid papulosis (2015).
================================================================
1.) Lymphomatoid papulosis associated with pregnancy.
2.) Lymphomatoid papulosis: successful weekly pulse super potent topical corticosteroid therapy in three pediatric patients.
3.) Lymphomatoid papulosis and cutaneous CD30+ lymphoma.
4.) Amplification of genomic DNA demonstrates the presence of the t(2;5) (p23;q35) in anaplastic large cell lymphoma, but not in other non-Hodgkin's lymphomas, Hodgkin's disease, or lymphomatoid papulosis [see comments]
5.) A case of lymphomatoid papulosis occurred simultaneously with Ki-1-positive anaplastic large cell lymphoma.
6.) Lymphomatoid papulosis type A: clinical, morphologic, and immunophenotypic study.
7.) Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis.
8.) Lymphomatoid papulosis in association with mycosis fungoides: a study of 15 cases.
9.) Involvement of the tongue by lymphomatoid papulosis.
10.) Lymphomatoid papulosis treated with extracorporeal photochemotherapy.
11.) Absence of anaplastic lymphoma kinase (ALK) and Epstein-Barr virus gene products in primary cutaneous anaplastic large cell lymphoma and lymphomatoid papulosis.
12.) Absence of Epstein-Barr virus in lymphomatoid papulosis. An immunohistochemical and in situ hybridization study [see comments]
13.) Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders.
14.) Follicular lymphomatoid papulosis.
15.) Is it lymphoma or lymphomatoid papulosis?
16.) Lymphomatoid papulosis: a low-grade T-cell lymphoma?
17.) The dominant T cell clone is present in multiple regressing skin lesions and associated T cell lymphomas of patients with lymphomatoid papulosis.
18.) The cytokine mRNA expression in primary cutaneous CD30-positive lymphoproliferative disorders: successful treatment with recombinant interferon-gamma.
19.) Low incidence of Epstein-Barr virus presence in primary cutaneous T-cell lymphoproliferations.
20.) Clonal disease in extracutaneous compartments in cutaneous T-cell lymphomas. A comparative study between cutaneous T-cell lymphomas and pseudo lymphomas.
21.) Apoptotic and proliferating cells in cutaneous lymphoproliferative diseases.
22.) No evidence of HTLV-I proviral integration in lymphoproliferative disorders associated with cutaneous T-cell lymphoma.
23.) Interleukin-7 receptor expression in cutaneous T-cell lymphomas.
24.) The t(2;5) in human lymphomas.
25.) Pityriasis lichenoides in children: clinicopathologic review of 22 patients.
26.) Regional lymphomatoid papulosis: a report of four cases.
27.) Infection with parapoxvirus induces CD30-positive cutaneous infiltrates in humans.
28.) CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: A clue to the pathophysiology of clinical regression.
29.) Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis.
30.) Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. A distinct clinicopathological entity with an aggressive clinical behavior.
31.) Lymphomatoid papulosis in association with mycosis fungoides: a study of 15 cases.
32.) Early detection of cutaneous lymphoma.
33.) Cutaneous pseudolymphomas.
34.) Lymphomatoid papulosis in childhood with exclusive acral involvement.
35.) Absence of HTLV-1 proviral sequences in patients with lymphomatoid papulosis.
36.) Clinicopathologic manifestations of Epstein-Barr virus-associated cutaneous lymphoproliferative disorders.
37.) Follicular lymphomatoid papulosis and multiple myeloma.
38.) Lymphomatoid papulosis--treatment with recombinant interferon alfa-2a and etretinate.
39.) Lymphomatoid papulosis: a follow-up study of 41 patients.
40.) Lymphomatoid papulosis associated with acquired ichthyosis.
41.) Primary anaplastic large-cell lymphoma of the skin. A case report suggesting that regressing atypical histiocytosis and lymphomatoid papulosis are subsets.
42.) Lymphomatoid papulosis: a clinical and histopathologic review of 53 cases with leukocyte immunophenotyping, DNA flow cytometry, and T-cell receptor gene rearrangement studies.
43.) [Lymphomatoid papulosis and anaplastic giant-cell lymphoma]
44.) Molecular evidence for a clonal relationship between lymphomatoid papulosis and Ki-1 positive large cell anaplastic lymphoma.
45.) Lymphomatoid papulosis followed by Hodgkin's lymphoma. Differential response to therapy [see comments]
46.) Epidemiology of lymphomatoid papulosis [see comments]
47.) Lethal midline granuloma (peripheral T-cell lymphoma) after lymphomatoid papulosis.
48.) The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas.
49.) Immunophenotypic and genotypic characterization of lymphomatoid papulosis.
50.) Pityriasis lichenoides and lymphomatoid papulosis.
51.) Lymphomatoid papulosis: clinicopathological comparative study with pityriasis lichenoides et varioliformis acuta.
52.) PUVA-induced lymphomatoid papulosis in a patient with mycosis fungoides.
53.) Accuracy in diagnosis of lymphomatoid papulosis.
54.) Regressing atypical histiocytosis and lymphomatoid papulosis: variants of the same disorder?
55.) Lymphomatoid papulosis and its relationship to "idiopathic" hypereosinophilic syndrome.
56.) Lymphomatoid papulosis/pityriasis lichenoides in two children.
57.) [Acyclovir in mycosis fungoides and lymphomatoid papulosis]
58.) Eosinophilic histiocytosis: a variant form of lymphomatoid papulosis or a disease sui generis?
59.) Lymphomatoid papulosis: a premalignant T cell disorder.
60.) Clinical and histologic differentiation between lymphomatoid papulosis and pityriasis lichenoides.
61.) The clinicopathologic spectrum of lymphomatoid papulosis: study of 31 cases.
62.) [Lymphomatoid papulosis in a child]
63.) Lymphomatoid papulosis in an 11-month-old infant.
64.) Topical carmustine therapy for lymphomatoid papulosis.
65.) Immunohistology of pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica. Evidence for their interrelationship with lymphomatoid papulosis.
====================================================
1.) Lymphomatoid papulosis associated with pregnancy.
====================================================
Author Yamamoto O; Tajiri M; Asahi M Address Department of Dermatology and Occupational Dermatopathology,
University of Occupational and Environmental Health, Kitakyushu, Japan.
Source Clin Exp Dermatol, 22(3):141-3 1997 May
Abstract
We report a case of lymphomatoid papulosis which developed in a 29-year-old pregnant woman. She had numerous papules scattered over the inner aspect of the left thigh. Histology of the biopsy specimen demonstrated an atypical mononuclear cell infiltration of the dermis. Spontaneous regression of the lesions occurred after termination of gestation. A possible effect of hormonal changes and alterations in T lymphocyte activity during pregnancy on the occurrence of lymphomatoid papulosis is discussed. In 1968, Macaulay introduced the term lymphomatoid papulosis for a chronic self-healing skin lesion which was clinically benign and histologically malignant. Clinically, lymphomatoid papulosis consists of involuting and recurring papules, plaques and nodules. Histopathologically, the lesion is characterized by an atypical lymphoid infiltrate which resembles malignant lymphoma. Immunohistochemically, the atypical lymphoid cells bear T-cell markers and are characterized by the expression of Ki-1 or CD30. We describe the first case of typical lymphomatoid papulosis which developed during pregnancy.
============================================================
2.) Lymphomatoid papulosis: successful weekly pulse super potent topical corticosteroid therapy in three pediatric patients.
============================================================
Author Paul MA; Krowchuk DP; Hitchcock MG; Jorizzo JL A
ddress Department of Dermatology, Bowman Gray School of Medicine, Wake Forest University, Winston-Salem, NC 27157, USA. Source Pediatr Dermatol, 13(6):501-6 1996 Nov-Dec
Abstract Lymphomatoid papulosis is a T-cell proliferation that occurs primarily in adults but has been well described in children. Lesions may regress spontaneously but often leave residual scarring and, as a result, intervention frequently is considered. Therapeutic modalities commonly employed for adults with lymphomatoid papulosis may be poorly tolerated by pediatric patients. We present a series of three children with lymphomatoid papulosis treated with superpotent topical corticosteroids (halobetasol or clobetasol propionate).
When applied twice daily for 2 to 3 weeks followed by weekly pulsed application, this treatment resulted in complete resolution of nearly all cutaneous lesions. Three ulcerated lesions, occurring in two patients, required adjuvant therapy with intralesional triamcinolone. To date one patient remains free of cutaneous disease and two children experience occasional new lesions that respond to renewed treatment with topical clobetasol propionate. None of the children have evidence of systemic disease. We conclude that pulsed application of a superpotent topical corticosteroid is efficacious and safe in the management of cutaneous lesions of lymphomatoid papulosis and avoids the risks often associated with more aggressive interventions. Since these agents do not alter the risk of subsequent malignancy, careful ongoing surveillance of children with lymphomatoid papulosis is imperative.
============================================================
3.) Lymphomatoid papulosis and cutaneous CD30+ lymphoma.
============================================================
Author LeBoit PE Address Department of Pathology, University of California, San Francisco 9
4143-0506, USA. Source Am J Dermatopathol, 18(3):221-35 1996 Jun
Abstract
Lymphomatoid papulosis and cutaneous CD30+ lymphoma are closely related conditions in which large atypical lymphocytes that have similar immunophenotypic features occur. In lymphomatoid papulosis, the lesions are papules and nodules that spontaneously involute. There are two polar histologic patterns, type A and B, in which the large atypical cells resemble those of Hodgkin's disease and mycosis fungoides, respectively, but in many cases, features of both types are present, either separately or in the same lesions. Variants of lymphomatoid papulosis include cases with a perifollicular distribution and those with lymphocytic vasculitis or dermal mucin deposits. Clinical lesions that tend to be stable, a monomorphous cellular composition, and in the case of immunocompromised patients, the presence of Epstein-Barr viral genome characterize cutaneous CD30+ lymphoma. A loss of response to transforming growth factor-beta, which normally dampens cellular proliferation, may differentiate CD30+ lymphoma from lymphomatoid papulosis.
============================================================
4.) Amplification of genomic DNA demonstrates the presence of the t(2;5) (p23;q35) in anaplastic large cell lymphoma, but not in other non-Hodgkin's lymphomas, Hodgkin's disease, or lymphomatoid papulosis [see comments]
============================================================
Author Sarris AH; Luthra R; Papadimitracopoulou V; Waasdorp M; Dimopoulos MA; McBride JA; Cabanillas F; Duvic M; Deisseroth A; Morris SW; Pugh WC Address Department of Hematology, University of Texas M.D. Anderson Cancer Center, Houston 77030, USA.
Source Blood, 88(5):1771-9 1996 Sep 1
Abstract
Anaplastic large cell lymphoma (ALCL) is a distinct clinicopathologic variant of intermediate grade non-Hodgkin's lymphomas (NHL) composed of large pleomorphic cells that usually express the CD30 antigen and interleukin (IL)-2 receptors, and is characterized by frequent cutaneous and extranodal involvement. With variable frequency ALCL bear the t(2;5)(p23;q35) chromosomal translocation that fuses the nucleophosmin (NPM) gene on chromosome 5q35 to a novel protein kinase gene, Anaplastic Lymphoma Kinase (ALK), on chromosome 2p23. We determined the frequency of this translocation with a novel DNA polymerase chain reaction (PCR) technique using 0.5 microgram of genomic DNA, 5'-primers derived from the NPM gene and 3'-primers derived from the ALK gene and hybridization with internal probes.
The presence of amplifiable DNA in the samples was tested with the inclusion in the PCR reaction of oligonucleotide primers designed to amplify a 3016-bp fragment from the beta-globin locus. NMP-ALK fusion amplicons were detected using DNA isolated either from all three ALCL cell lines tested, or from all four primary ALCL tumors known to contain the t(2;5)(p23;q35) translocation. Nested amplicons were detected by hybridization in 100% of specimens diluted 10(4)-fold and in 20% of those diluted 10(5)-fold. We subsequently examined archival genomic DNA from 20 patients with ALCL, 39 with diffuse large cell, 2 with mantle cell, 20 with peripheral T cell, 13 with low-grade NHL, 31 with Hodgkin's disease (HD), and 6 with lymphomatoid papulosis. Fusion of the NPM and ALK genes was detected in three of 18 patients with ALCL who had amplifiable DNA (17%, 95% confidence intervals 4% to 41%), but not in any patients with other NHL, HD, or lymphomatoid papulosis.
The amplicon sizes were different in all cell lines and patients reflecting unique genomic DNA breakpoints. We conclude that with genomic DNA-PCR the rearrangement of the NPM and ALK loci is restricted to patients with ALCL. Further studies are needed to determine the prognostic significance of the NPM-ALK rearrangement, to determine whether its detection can aid in the differential diagnosis between ALCL. Hodgkin's disease, and lymphomatoid papulosis, and to establish the usefulness of the genomic DNA PCR in the monitoring of minimal residual disease in those patients whose tumors bear the t(2;5).
============================================================
5.) A case of lymphomatoid papulosis occurred simultaneously with Ki-1-positive anaplastic large cell lymphoma.
============================================================
Author Lee NS; Cha SW; Hong SJ; Shin WY; Lee GT; Jeon JW; Won JH; Baick SH; Hong DS; Park HS Address Department of Internal Medicine, Soonchunhyang University, College of Medicine, Seoul, Korea.
Source Korean J Intern Med, 12(1):84-8 1997 Jan
Abstract
Lymphomatoid papulosis (LyP) is a chronic self-healing skin eruption that is clinically benign but histologically mimics a malignant lymphoma. However, lymphomatoid papulosis with anaplastic large cell lymphoma responds poorly to medical treatments, including chemotherapies. We experienced a 60-year-old male patient with lymphomatoid papulosis occurred simultaneously with relapsed Ki-1-positive anaplastic large cell lymphoma who was treated with salvage chemotherapy but, unfortunately, failed to be rescued. We report it with a review of the literature.
============================================================
6.) Lymphomatoid papulosis type A: clinical, morphologic, and immunophenotypic study.
============================================================
Author /br Sioutos N; Asvesti C; Sivridis E; Aygerinou G; Tsega A; Zakopoulou N; /br Zographakis I Address Department of Pathology, Georgetown University, Washington, DC, USA.
Source Int J Dermatol, 36(7):514-7 1997 Jul
Abstract
BACKGROUND: Lymphomatoid papulosis (LyP) is a cutaneous clonal or polyclonal Ki-1 + T-cell lymphoproliferative disorder, morphologically resembling Ki-1 + anaplastic large cell lymphomas (Ki-1 + ALCL) or Hodgkin's disease (HD). Lymphomatoid papulosis usually has a characteristic benign clinical course with remissions and relapses of the cutaneous eruptions.
METHODS: The authors studied three patients with LyP. In each case the diagnosis was established based on the typical clinical history and presentation of the cutaneous lesions as well as the morphologic and immunophenotypic findings.
RESULTS: In all three cases the skin biopsies showed a polymorphic, nonepidermotropic, dermal lymphocytic infiltrate, composed of small lymphocytes and fewer large, atypical cells. The large cells were positive for the activation markers CD30 (Ki-1) and CD45R (leukocyte common antigen), and were negative for the HD marker CD15 (Leu MI).
CONCLUSIONS: In most cases, LyP can be distinguished from Ki-1 + ALCL and HD on the basis of clinical, morphologic, and/or immunophenotypic findings. We emphasize the importance of the recognition of LyP as a clinicopathologic entity and the awareness of dermatologists, oncologists, and surgical pathologists in differentiating LyP from other primary cutaneous Ki-1 + lymphoproliferative disorders (Ki-1 + ALCL and HD). The prognosis of cutaneous Ki-1 + ALCL and HD is usually different from LyP and requires a different therapeutic approach.
============================================================
7.) Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis.
============================================================
Author Wang HH; Myers T; Lach LJ; Hsieh CC; Kadin ME Address Department of Pathology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts 02115, USA.
Source Cancer, 86(7):1240-5 1999 Oct 1
Abstract
BACKGROUND: Lymphomatoid papulosis (LyP) is a rare skin disease with malignant potential. The long term outcomes of patients with this disease have not been adequately assessed.
METHODS: Fifty-seven patients with biopsy-proven LyP and 67 controls matched for age, gender, and race were followed prospectively from 1988 to 1996. Reported malignancies were confirmed by surgical pathology and/or autopsy reports. A search through the National Death Index through December 1995 was conducted to identify all deaths, and death certificates were procured. Expected numbers of malignancies based on SEER data were calculated for both the patient and the control groups.
RESULTS: Six LyP patients (10.5%) and 1 control (1.5%) reported nonlymphoid malignancies (P = 0.047). Two patients and no controls developed lymphoid malignancies (mycosis fungoides and CD30(+) cutaneous lymphoma). The expected numbers of nonlymphoid and lymphoid malignancies in the LyP patient group, based on the SEER data, were 1.93 and 0.15, respectively, yielding a relative risk (with 95% confidence interval) of 3.11 (1.26-6.47) for nonlymphoid malignancies and 13.33 (2.24-44.05) for malignant lymphomas in the LyP patients. There was no significant difference between the observed and expected numbers of malignancies in the control group. Four LyP patients died during the follow-up, three due to malignancies; and one control died of a gunshot wound to the head (suicide). The difference in overall survival between the LyP patients and the controls was not statistically significant (P = 0. 12).
CONCLUSIONS: Patients with LyP appear to have an increased risk of both lymphoid and nonlymphoid malignancies. The increased risk of nonlymphoid as well as lymphoid malignancies may suggest a basic underlying genetic defect leading to the development of malignancy in LyP patients. Copyright 1999 American Cancer Society.
============================================================
8.) Lymphomatoid papulosis in association with mycosis fungoides: a study of 15 cases.
============================================================
Author Basarab T; Fraser-Andrews EA; Orchard G; Whittaker S; Russel-Jones R Address St John's Institute of Dermatology, St Thomas' Hospital, London, UK.
Source Br J Dermatol, 139(4):630-8 1998 Oct
Abstract
We report clinical findings in 15 patients with lymphomatoid papulosis (LyP) associated with mycosis fungoides (MF). LyP either preceded (n = 4), followed (n = 5) or occurred concurrently with the MF lesions (n = 6). Twenty-eight LyP lesions were classified histologically and analysed further with immunostaining for CD3 and CD30. Five biopsies contained a predominance of type A cells, six biopsies contained a predominance of type B cells. and six were mixed (A + B). However, 11 biopsies contained a population of atypical mononuclear cells with large hyperchromatic nuclei that we have termed indeterminate cells.
These cells contained a thin rim of eosinophilic cytoplasm and showed strong CD30 but absent, faint or normal CD3 staining. In seven biopsies from five separate patients these cells represented the predominant cell type and we have termed this the pleomorphic variant of LyP. Analysis of T-cell receptor genes using Southern blot analysis and polymerase chain reaction/single strand conformational polymorphism analysis identified a T-cell clone in six of 16 LyP lesions and nine of 16 MF lesions. In the three patients who had clones in both types of skin lesions, the clones were identical. Only two of 10 blood samples, both of which were from the same patient, had a T-cell clone and none of two lymph nodes showed evidence of a clonal population. To date all patients are alive with a median follow-up of 15 years from the onset of the first lesion. One patient has developed Lin anaplastic large cell lymphoma of the nasopharynx. These data augment the current literature on the association of LyP and MF and suggest that the
============================================================
9.) Involvement of the tongue by lymphomatoid papulosis.
============================================================
Author Kato N; Tomita Y; Yoshida K; Hisai H Address Department of Dermatology and Research Institute, National Sapporo Hospital, Japan.
Source Am J Dermatopathol, 20(5):522-6 1998 Oct
Abstract
We report on a case of lymphomatoid papulosis (LyP) with involvement of the tongue. The patient was a 34-year-old Japanese man. Three reddish, centrally depressed, slightly elevated nodules were evident on the dorsal tongue, along with lesions elsewhere on the skin. One of them was biopsied and exhibited a superficial and deep, perivascular and interstitial mixed cellular infiltrate including atypical lymphoid cells, lymphocytes, neutrophils, and histiocytes.
The patient also showed rhythmical recurrence of reddish papules and ulcerated nodules on the trunk, extremities, and anogenital area. Histologically, these papules showed a dense, wedge-shaped mixed cellular infiltrate in the dermis, which included medium and large atypical lymphoid cells, lymphocytes, neutrophils, and histiocytes. Immunoperoxidase staining for CD30 was positive in the cell membrane and cytoplasm of the atypical cells. We could not find other reports of LyP involving the tongue. Systemic treatment with interferon (INF)-alpha2a was dramatically effective in inhibiting recurrence of the eruption.
============================================================
10.) Lymphomatoid papulosis treated with extracorporeal photochemotherapy.
============================================================
Author Wollina U Address Department of Dermatology, University of Jena, 07740 Jena, Germany. Source Oncol Rep, 5(1):57-9 1998 Jan-Feb
Abstract
Lymphomatoid papulosis (LP) is a rare low-grade T-cell lymphoma which may respond to cytotoxic drugs and PUVA irradiation but long-term remission has not been achieved. Extracorporeal photochemotherapy (ECP) is an immunomodulating therapy used successfully for several types of CTCL, no experience with LP has been reported yet. ECP therapy with 8-methoxypsoralen was introduced on two subsequent days once per month for half a year in a 42-year old women with a 20-year history of LP. Disseminated papules disappeared rapidly after 3 cycles of ECP treatment but metastatic spread continued, which made necessary a subsequent polychemotherapy. One year later, the patient died of central nervous system metastasis. ECP monotherapy seems unable to control disease progression in LP despite beneficial effects on skin lesions.
============================================================
11.) Absence of anaplastic lymphoma kinase (ALK) and Epstein-Barr virus gene products in primary cutaneous anaplastic large cell lymphoma and lymphomatoid papulosis.
============================================================
Author Herbst H; Sander C; Tronnier M; Kutzner H; H¨ugel H; Kaudewitz P Address Institut f¨ur Pathologie, Universit¨atsklinikum Benjamin Franklin, Freie Universit¨at, Berlin, Germany. Source Br J Dermatol, 137(5):680-6 1997 Nov
Abstract
The prevalence of the t(2;5)(p23;q35) and/or anaplastic lymphoma kinase (ALK) gene products in cutaneous anaplastic large cell (ALC) lymphomas and a potential precursor lesion, lymphomatoid papulosis (LyP), is controversial. ALK gene products, which are absent from normal lymphohaematopoietic cells, are a phenotypic marker of lymphomas carrying the t(2;5). We used in situ hybridization and immunohistology to screen 14 cutaneous ALC lymphomas, 21 cases of LyP, and one nodal ALC lymphoma associated with LyP for ALK gene products. ALK gene products were not detectable in these cases. In contrast, ALK gene products were found in a lymphonodal ALC lymphoma with subsequent extension to the skin and in t(2;5)-positive cell lines. Detection of the Epstein-Barr virus (EBV)-encoded small nuclear transcripts (EBER), and of immunoglobulin light chain transcripts served to check for the presence of cellular RNA in the tissue sections. EBER transcripts were found in scattered reactive lymphoid cells, but not in atypical or tumour cells. ALK gene expression and EBV infection seem to be a rare finding in cutaneous ALC lymphomas and LyP. This points to a molecular aetiology of primary cutaneous ALC lymphomas and LyP distinct from that of extracutaneous CD30+ lymphoproliferative disease. Detection of the t(2;5) or ALK gene products in cutaneous lymphoproliferative lesions therefore requires exclusion of extracutaneous ALC lymphoma in such patients.
============================================================
12.) Absence of Epstein-Barr virus in lymphomatoid papulosis. An immunohistochemical and in situ hybridization study [see comments]
============================================================
Author Sang¨ueza OP; Galloway J; Eagan PA; Braziel RM; Gulley ML Address Department of Pathology and Medicine, Medical College of Georgia, Augusta, USA.
Source Arch Dermatol, 132(3):279-82 1996 Mar
Abstract
BACKGROUND AND DESIGN: Lymphomatoid papulosis (LyP) and cutaneous Hodgkin's disease share many clinical, histopathologic, and immunohistochemical features. Epstein-Barr virus (EBV) has been implicated in the pathogenesis of several lymphoid malignancies, including Hodgkin's disease. Given the similarities between LyP and Hodgkin's disease, we asked if EBV could be detected in lesions of LyP. We examined 31 specimens of LyP that were obtained from 24 patients for evidence of EBV by in situ hybridization to EBER1 transcripts and for immunohistochemistry of viral latent membrane protein 1 (LMP1).
RESULTS: In no instance there was there any evidence of EBV gene products by either in situ hybridization or immunohistochemistry.
CONCLUSIONS: The absence of EBV in LyP suggests that this virus is not operative in the pathogenesis of LyP. Furthermore, it suggests that LyP and Hodgkin's disease may not share the same molecular mechanisms despite their phenotypic similarities.
============================================================
13.) Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders.
============================================================
Author Vonderheid EC; Sajjadian A; Kadin ME Address Department of Dermatology, Medical College of Pennsylvania, Philadelphia, USA.
Source J Am Acad Dermatol, 34(3):470-81 1996 Mar
Abstract
BACKGROUND: The spectrum of primary cutaneous CD30+ lymphoproliferative disease consists of lymphomatoid papulosis (LyP) at one extreme and CD30+ peripheral T-cell lymphoma (Ki-1+ lymphoma) presenting in the skin at the other extreme. Methotrexate has been reported to be effective in LyP, but the experience has been limited to single case reports or small series. OBJECTIVE: The objective was to determine the effectiveness of methotrexate in the treatment of primary cutaneous DC30+ lymphoproliferative disease.
METHODS: We reviewed our 20-year experience with the use of methotrexate in 45 patients with relatively severe LyP, Ki-1+ lymphoma, and interface presentations.
RESULTS: During induction of methotrexate therapy patients received maximum doses ranging from 10 to 60 mg/week (median, 20 mg/week). Clinical improvement usually occurred quickly, typically at doses of 15 to 20 mg weekly, and satisfactory long-term control was achieved in 39 patients (87%) with maintainance doses given at 10 to 14-day intervals (range, 7 to 28 days). After methotrexate was discontinued, 10 patients remained free of CD30+ lesions from more than 24 months to more than 227 months (median, more than 127 months). The median total duration of methotrexate therapy for all patients exceeded 39 months (range, 2 to 205 months). Adverse effects were generally mild and transient and included fatigue (47%), nausea (22%), weight loss (13%), diarrhea or gastrointestinal cramping (10%), increased serum hepatic transaminase levels (27%), anemia (11%), or leukopenia (9%). Early hepatic fibrosis was found in 5 of 10 patients, all of whom had been treated for more than 3 years (range, 38 to 111 months).
CONCLUSION: Low-dose methotrexate (25 mg or less given at 1-to 4-week intervals) is an effective and well-tolerated treatment of selected patients with primary cutaneous CD30+ lymphoproliferative disease.
============================================================
14.) Follicular lymphomatoid papulosis.
============================================================
Author Kato N; Matsue K Address Department of Dermatology, Hokkaido University School of Medicine, Japan. Source Am J Dermatopathol, 19(2):189-96 1997 Apr
Abstract
A case of follicular lymphomatoid papulosis (LyP) is reported. The patient was a 60-year-old Japanese woman. Clinically, cutaneous eruptions were reddish, centrally depressed, dome-shaped papules on the extensor aspect of the forearm. Histologically, they exhibited features that fulfilled the disease criteria described by Pierard, et al., i.e., (Am J Dermatopathol 1980;2:173-80), mixed cellular infiltrates including atypical Reed-Sternberg cell-like type-A cells and mycosis cell-like type-B cells surrounding hyperplastic follicular epithelia. The patient also showed many typical nonfollicular LyP papules, i.e., rhythmically recurrent papules which underwent spontaneous involution within a few weeks, over a 10-year period. The coincidental occurrence of a rare variant of follicular LyP and typical LyP in the same individual further suggests that follicular LyP is merely a histological pattern of LyP involving epithelial adnexae.
============================================================
15.) Is it lymphoma or lymphomatoid papulosis?
============================================================
Author Demierre MF; Goldberg LJ; Kadin ME; Koh HK Address Department of Dermatology, Boston University School of Medicine, MA, USA.
Source J Am Acad Dermatol, 36(5 Pt 1):765-72 1997 May
Abstract
Distinguishing malignancy from premalignant conditions can be difficult. Controversy surrounds both the clinical and histologic criteria used to distinguish lymphomatoid papulosis, a benign disorder, from CD30+ anaplastic large-cell lymphoma. Three case histories illustrate important points in categorizing different lymphoproliferative disorders as benign or malignant. We emphasize a multidisciplinary approach to improve diagnosis and patient management.
============================================================
16.) Lymphomatoid papulosis: a low-grade T-cell lymphoma?
============================================================
Author Orchard GE Address Dermatopathology Department, St Thomas' Hospital, London, England. Source Br J Biomed Sci, 53(2):162-9 1996 Jun
Abstract
Lymphomatoid papulosis is a chronic cutaneous lymphoid disease characterised clinically by the presence of recurrent papulonodular or plaque like lesions, which appear benign. Paradoxically, histological and cytopathological features demonstrate features of malignancy. This annotation highlight the current theories and technical advances into the assessment of this condition, with emphasis on possible pathogenic disease mechanisms.
============================================================
17.) The dominant T cell clone is present in multiple regressing skin lesions and associated T cell lymphomas of patients with lymphomatoid papulosis.
============================================================
Author
Chott A; Vonderheid EC; Olbricht S; Miao NN; Balk SP; Kadin ME Address Department of Pathology, Beth Israel Hospital, Boston, Massachusetts, USA.
Source J Invest Dermatol, 106(4):696-700 1996 Apr
Abstract
This study was undertaken to determine the clonality of lymphomatoid papulosis (LyP), its clonal relationship to lymphomas, which occur at high frequency in LyP patients, and to define the cell lineage of Reed-Sternberg-like cells in type A lesions of LyP. Punch biopsies of skin of 11 adult patients with LyP were analyzed for morphologic subtype of LyP, surface antigens, and clonal T-cell receptor (TCR) gene rearrangements. Clonal rearrangements were identified by semiquantitative polymerase chain reaction amplification and sequencing of TCR-beta chain genes in nine patients and TCR-gamma chain genes in two patients.
A single dominant clone was detected in multiple separate LyP lesions, often of different histologies, in nine patients. The same clone was detected in LyP lesions and the anaplastic large cell lymphoma (ALCL) of 2 patients and the mycosis fungoides (MF) of 2 other patients. No dominant clone could be detected in one patient with LyP uncomplicated by lymphoma or in a second patient with LyP and MF. A T-cell lineage was evident for RS-like cells in cell culture and in type A lesions. These results show that multiple regressing skin lesions and associated T cell lymphomas (MF and ALCL) are clonally related in most LyP patients, which suggest that the disease in these patients was initiated by a non-random genetic event.
============================================================
18.) The cytokine mRNA expression in primary cutaneous CD30-positive lymphoproliferative disorders: successful treatment with recombinant interferon-gamma.
============================================================
Author Yagi H; Tokura Y; Furukawa F; Takigawa M Address Department of Dermatology, Hamamatsu University School of Medicine, Japan. S
ource J Invest Dermatol, 107(6):827-32 1996 Dec
Abstract
Primary cutaneous CD30 (Ki-1)+ large cell lymphoma (KiL) and lymphomatoid papulosis (LyP) type A are collectively termed as primary cutaneous CD30-positive lymphoproliferative disorders. We examined the cytokine profile of skin-infiltrating cells and the therapeutic efficacy of recombinant interferon-gamma (rIFN-gamma) in primary cutaneous KiL and LyP type A. By reverse transcriptase-polymerase chain reaction, mRNAs for interleukin-4 (IL-4) and IL-10 were detected in the dermis of skin lesions in all cases (three cases of KiL and four cases of LyP).
In addition, tissue from one KiL patient transcribed IL-2 and IFN-gamma messages, and one LyP patient showed IL-2 mRNA. In contrast, normal skin from ten healthy donors contained mRNA for IL-2 or IFN-gamma, or both, but not for IL-4. Before the therapeutic trial of rIFN-gamma, the response of skin lesions was assessed by a predictive skin test with local injection of rIFN-gamma (0.5 x 10(6) Japan Reference Units [JRU; 1 JRU roughly corresponds to 4 NIH units]) for 3 consecutive days in two KiL and two LyP patients. Numbers of skin-infiltrating CD30+ cells were decreased, and transcription of mRNA for IL-4 and IL-10 was downregulated after the skin test in one KiL and two LyP cases.
One KiL patient showed no histologic response or change in mRNA expression. In the therapeutic trial, rIFN-gamma (total doses of 1.2-4.0 x 10(7) JRU) was administered intravenously (n = 2) or locally (n = 2). In three patients who responded to the skin test, the lesions were objectively improved and the numbers of skin-infiltrating CD30+ cells were markedly decreased after the therapeutic trial. No improvement was observed in one KiL patient who did not respond to the skin test. These findings suggest that the skin-infiltrating CD30+ cells in KiL and LyP have a Th2 cytokine profile and raise the possibility that the administration of rIFN-gamma improves the conditions by inhibiting cytokine mRNA transcription and proliferation of CD30+ cells.
============================================================
19.) Low incidence of Epstein-Barr virus presence in primary cutaneous T-cell lymphoproliferations.
============================================================
Author Anagnostopoulos I; Hummel M; Kaudewitz P; Korbjuhn P; Leoncini L; Stein H Address Klinikum Benjamin Franklin, Free University Berlin, Germany.
Source Br J Dermatol, 134(2):276-81 1996 Feb
Abstract
Multiple biopsies taken from 76 European human immunodeficiency virus (HIV)-negative patients with primary cutaneous T-cell lymphoproliferations, including mycosis fungoides (MF), pleomorphic T-cell lymphoma (PMTCL), anaplastic large cell lymphoma (ALCL) and lymphomatoid papulosis (LyP) were investigated for the presence of Epstein-Barr virus (EBV) through a combined approach. Polymerase chain reaction (PCR) was employed for EBV-DNA detection, in situ hybridization (ISH) for cellular localization of EBV-encoded nuclear RNAs (EBER1 and EBER2) and immediate early Bam H-fragment; lower frame (BHLF) RNA, and immunohistology (IH) for the identification of EBV-encoded latent membrane protein 1 (LMP1) and of nuclear antigen (EBNA) 2 expression. EBV-DNA was detectable by PCR in 15 of 76 cases (19.7%). EBER-ISH combined with IH identified a variable, usually very low, number of infected neoplastic cells in only seven of the 15 EBV-DNA-harbouring cases.
This discrepancy between the results obtained with PCR and ISH is apparently caused by the low number of the infected cells per tissue section. The PMTCL entity produced the greatest number of positive cases, whilst ALCL and LyP cases were almost constantly devoid of the virus. BHLF transcripts were not detectable in any case, nor did any of the EBER-positive cells show an LMP1 or EBNA2 expression. These data show that primary cutaneous T-cell lymphoproliferations display an infrequent association with a latent EBV infection and that the pathogenic role of the virus in the positive cases remains obscure as the virus frequently infects only a minority of the atypical cells.
============================================================
20.) Clonal disease in extracutaneous compartments in cutaneous T-cell lymphomas. A comparative study between cutaneous T-cell lymphomas and pseudo lymphomas.
============================================================
Author Dommann SN; Dommann-Scherrer CC; Dours-Zimmermann MT; Zimmermann DR; Kural-Serbes B; Burg G Address University Hospital of Z¨urich, Department of Dermatology, Switzerland. Source
Arch Dermatol Res, 288(4):163-7 1996 Apr
Abstract
Extracutaneous involvement is a sign of poor prognosis in cutaneous T-cell lymphomas (CTCL). Unfortunately it becomes clinically and histologically manifest only late in the course of the disease. It was the purpose of this study to detect clonality in peripheral blood, lymph nodes and bone marrow samples at times when extracutaneous involvement cannot otherwise be demonstrated. In addition to skin biopsies, peripheral blood, lymph node and bone marrow samples from a total of 25 patients were analysed by Southern blotting for clonal gene rearrangement of the T-cell receptor beta-chain.
Six of the patients were suffering from mycosis fungoides (MF), four from non-MF CTCL (pleomorphic T-cell lymphomas), seven from S´ezary syndrome (SS), eight from pseudolymphoma (insect bites) (PSL), and one from lymphomatoid papulosis (LP). Clonal TcR b gene rearrangements were found in patients with MF in four of five skin probes as well as in two of two lymph node samples and in one of two peripheral blood samples.
In SS patients, all skin probes (seven of seven), lymph node samples (six of six), peripheral blood samples (six of six) and one bone marrow specimen had a clonal TcR beta gene rearrangement. In patients with non-MF CTCL, two of four skin, zero of two peripheral blood and one of one bone marrow samples with clonal T cells were detected. All investigated patients showed exactly the same rearrangement pattern at extranodal sites and in the skin, which is proof for the same clone in all compartments. In contrast, no rearrangements were detected in LP and PSL (zero of eight skin probes, zero of two peripheral blood samples).
Our results provide strong evidence for an early systemic spread of neoplastic cells in CTCL. However, an initial tumour burden has to be reached in order to lead to a clinically and prognostically relevant manifestation.
============================================================
21.) Apoptotic and proliferating cells in cutaneous lymphoproliferative diseases.
============================================================
Author Kikuchi A; Nishikawa T Address Department of Dermatology, Keio University School of Medicine, Tokyo, Japan.
Source Arch Dermatol, 133(7):829-33 1997 Jul
Abstract
BACKGROUND: The cell production vs the cell loss rate in one of the most important parameters in evaluating growth and biological behavior of neoplasms. Individual cell disintegration in tissues, apoptosis, is a constant finding in various tumors and has been shown, by using several techniques, as a recognizable cell death that is different from necrosis.
DESIGN: We studied the apoptosis-proliferation ratio in various lymphoproliferative disorders in the skin, including mycosis fungoides (MF), cutaneous T-cell lymphoma showing solid tumor mass (CTCL), B-cell lymphoma of the skin (BCL), lymphomatoid papulosis (LyP), and cutaneous pseudolymphoma by using terminal deoxyuridine triphosphate (dUTP)-biotin nick end labeling (TUNEL), a newly developed method to detect internucleosomal breaks characteristic of apoptotic cells.
SETTING: University referral center. PATIENTS: Fifty patients with cutaneous lymphoproliferative diseases.
MAIN OUTCOME MEASURES: Proliferation indexes and apoptosis index calculated by using immunohistochemical techniques.
RESULTS: The proliferation indexes in pseudolymphoma, which were calculated by using immunohistochemical analyses with anti-proliferating cell nuclear antigen and anti-MIB-1 monoclonal antibodies, were significantly lower than the indexes of MF, CTCL, BCL, and LyP, whereas, the apoptosis index in Lyp was significantly higher than in any other lymphoproliferative diseases studied. The apoptosis-proliferation ratio in the tumor stage of MF, CTCL, and BCL was almost constant, but the ratios in LyP and the plaque stage of MF were significantly higher than in the other diseases studied.
CONCLUSIONS: The clinical behavior of each lymphoproliferative disease in the skin seemed to be reflected in the apoptosis and proliferation indexes. We conclude that these indexes may become useful factors in the determination of the diagnosis and the prognosis for patients with lymphoproliferative diseases.
============================================================
22.) No evidence of HTLV-I proviral integration in lymphoproliferative disorders associated with cutaneous T-cell lymphoma.
============================================================
Author
Wood GS; Schaffer JM; Boni R; Dummer R; Burg G; Takeshita M; Kikuchi M Address Department of Dermatology, Case Western Reserve University, Cleveland, Ohio, USA.
Source Am J Pathol, 150(2):667-73 1997 Feb
Abstract
Several recent studies have reported detection of HTLV-I genetic sequences in patients with cutaneous T-cell lymphoma (CTCL) including mycosis fungoides and Sezary syndrome. The purpose of this study was to determine whether HTLV-I was detectable in lesional tissues of patients suffering from diseases known to be associated with CTCL. Thirty-five cases were obtained from diverse geographical locations including Ohio, California, Switzerland, and Japan. Six of them had concurrent CTCL. Cases were analyzed using a combination of genomic polymerase chain reaction (PCR)/ Southern blot, dot blot, and Southern blot analyses.
All assays were specific for HTLV-I provirus. Sensitivity ranged from approximately 10(-6) for PCR-based studies to 10(-2) for unamplified genomic blotting. Lesional DNA from patients with lymphomatoid papulosis (fourteen cases), Hodgkin's disease (twelve cases), and CD30+ large-cell lymphoma (nine cases) was tested for the HTLV-I proviral pX region using a genomic PCR assay followed by confirmatory Southern blot analysis with a nested oligonucleotide pX probe. All cases were uniformly negative.
All of the Hodgkin's disease cases, eight of the large-cell lymphoma cases, and six of the lymphomatoid papulosis cases were then subjected to dot blot analysis of genomic DNA using a full-length HTLV-I proviral DNA probe that spans all regions of the HTLV-I genome. Again, all cases were negative. Finally, eleven of the Hodgkin's disease cases were also subjected to Southern blot analysis of EcoRI-digested genomic DNA using the same full-length HTLV-I probe. Once again, all cases were negative. These findings indicated that, despite utilization of a variety of sensitive and specific molecular biological methods, HTLV-I genetic sequences were not detectable in patients with CTCL-associated lymphoproliferative disorders. These results strongly suggest that the HTLV-I retrovirus is not involved in the pathogenesis of these diseases.
============================================================
23.) Interleukin-7 receptor expression in cutaneous T-cell lymphomas.
============================================================
Author Bagot M; Charue D; Boulland ML; Gaulard P; Revuz J; Schmitt C; Wechsler J Address Department of Dermatology, University Paris XII, H^opital Henri Mondor, Cr´eteil, France.
Source Br J Dermatol, 135(4):572-5 1996 Oct
Abstract
Keratinocyte-derived interleukin-7 (IL-) is a potent growth factor for some cutaneous T-cell lymphomas (CTCL). We investigated the expression of IL-7 receptor (IL-7R) in several types of cutaneous and nodal lymphomas. We studied 44 CTCL (13 mycosis fungoides, six S´ezary syndromes, eight pleomorphic small cell, and 17 pleomorphic medium and large cell), 10 lymphomatoid papulosis (LP), five cutaneous B-cell lymphomas, and five reactive lymphocytic infiltrates. Twenty nodal T-cell lymphomas, and three reactive lymph nodes were also analysed. Frozen sections were stained with monoclonal antibodies directed against IL-7R, CD25, CD30 and T antigens (CD3, CD2, CD5, CD7, CD4, CD8), using the alkaline phosphatase-antialkaline phosphatase technique.
No expression of IL-7R was observed in cutaneous B-cell lymphomas, benign cutaneous lymphoid infiltrates, and reactive lymph nodes. IL-7R was expressed by more than 20% of lymphoid cells in 50-75% of all histological subtypes of CTCL, and by more than 50% of cells in 15-50%. IL-7R was expressed by more than 20% and 50% of cells in 40% and 10% of nodal large T-cell lymphomas, respectively. Eighty-nine per cent of CTCL and LP expressing IL7-R also expressed CD25+, compared with 58% of IL-7R--CTCL and LP (P < 0.05). No association of IL7-R and CD30 expression was found. In conclusion, CTCL frequently express IL-7R. This expression is not related to the epidermotropic characteristic of the infiltrate. In CTCL and LP, IL-7R expression is associated to CD25 expression, but not to CD30 expression.
============================================================
24.) The t(2;5) in human lymphomas.
============================================================
Author
Kadin ME; Morris SW Address Department of Pathology, Beth Israel Hospital and Harvard Medical School, Boston, MA 02115, USA. mkadin@bidmc.harvard.edu
Source Leuk Lymphoma, 29(3-4):249-56 1998 Apr
Abstract
A recurrent, reciprocal balanced translocation, t(2;5) (p23;q35), has been recognized in CD30+ anaplastic large-cell lymphomas (ALCL), a newly recognized subtype comprising approximately 5% of all non-Hodgkin's lymphoma (NHL). This translocation creates a novel fusion protein, NPM-ALK, which has transforming properties in vitro and can cause large-cell lymphoma in vivo when transfected into murine bone marrow.
Multiple techniques including reverse transcriptase-polymerase chain reaction (RT-PCR) amplification of NPM-ALK fusion transcripts, genomic DNA-PCR, RNA in situ hybridization, and fluorescence in situ hybridization (FISH) of metaphase chromosomes and interphase nuclei, and immunohistochemical detection of the 80 kilodalton protein (p80) derived from the NPM-ALK fusion have enabled surveys of normal and lymphoma tissues for evidence of the translocation. These studies suggest that expression of ALK protein, a novel orphan receptor tyrosine kinase, is normally confined to the nervous system. In lymphoma, NPM-ALK expression is most often seen in young patients with the monomorphic or small-cell variant of ALCL who present with advanced stage disease and have tumors with a CD30+, T- or null-cell phenotype.
It is less frequently detected in older patients and in ALCL of pleomorphic histology. In addition, expression of NPM-ALK has been found in occasional CD30 negative B-cell lymphomas with diffuse large cell or immunoblastic histology. NPM-ALK is rarely, if ever, detected in Hodgkin's disease or secondary ALCL. Although initially found in primary nodal ALCL, recent studies suggest that NPM-ALK expression may occur in lymphoma at extranodal sites, including the skin; it remains controversial, however, whether CD30+ primary cutaneous lymphoma and its benign counterpart, lymphomatoid papulosis (LyP), express NPM-ALK in some cases.
A retrospective study has suggested that expression of NPM-ALK is associated with a better overall 5-year survival; these results must be confirmed in prospective studies of patients with uniform staging and therapy to more fully understand the clinical significance of the t(2;5) and its novel chimeric protein, NPM-ALK.
============================================================
25.) Pityriasis lichenoides in children: clinicopathologic review of 22 patients.
============================================================
Author Roman´i J; Puig L; Fern´andez-Figueras MT; de Moragas JM Address Department of Dermatology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain.
Source Pediatr Dermatol, 15(1):1-6 1998 Jan-Feb
Abstract
Pityriasis lichenoides (PL) is a cutaneous disease of unknown origin, with an autoinvolutive course, that can occur in pediatric patients. Traditionally, acute and chronic variants have been described, but other special forms of presentation have been reported.
We reviewed the clinical records and histopathologic specimens of all pediatric patients diagnosed with PL in our hospital from 1980 to 1995 to assess the clinicopathologic features of this disorder in our environment. Twenty-two of the 118 cases reviewed were pediatric patients less than 15 years old (12 males and 10 females, 18.6% of all patients). Their ages ranged from 3 to 15 years, with a mean of 9.3 years.
Most of the patients (72%) had the chronic variant of the disease, while the remainder had an acute course. One patient suffered from acute ulceronecrotic PL. Systemic treatments prescribed were erythromycin in eight patients, PUVA in five patients, and methotrexate in one patient. Three patients had a prolonged course with more than two episodes. Acute and chronic PL are polar extremes, but individual cases cannot be classified only on the basis of histopathologic data, since coexistence of lesions in different stages of evolution can lead to sampling bias.
Acute ulceronecrotic forms and the presence of a variable degree of cellular atypia in the infiltrate are liable to cause differential diagnostic problems with lymphomatoid papulosis (LP), which cannot be completely resolved on the basis of T-cell receptor clonal rearrangement detection.
============================================================
26.) Regional lymphomatoid papulosis: a report of four cases.
============================================================
Br J Dermatol 1999 Dec;141(6):1125-1128
Scarisbrick JJ, Evans AV, Woolford AJ, Black MM, Russell-Jones R
Lymphomatoid papulosis (LyP) is a chronic self-healing cutaneous eruption which is clinically benign but histologically malignant. Lesions occur episodically over the trunk and limbs. We describe four patients with regional LyP. All were male, with a range in age at onset from 12 to 47 years. In all cases, lesions were confined to a segmental unilateral area. Two patients had type A and two type B LyP. We have long-term follow-up on one patient whose lesions were limited to the right buttock for more than 20 years before more widespread lesions developed. Another patient with lesions on the left flank had mycosis fungoides limited to the same region. Only one other case of LyP presenting in a regional distribution has previously been described.
============================================================
27.) Infection with parapoxvirus induces CD30-positive cutaneous infiltrates in humans.
============================================================
J Cutan Pathol 1999 Nov;26(10):520-2
Rose C, Starostik P, Brocker EB Department of Dermatology, University of Wurzburg, Germany.
Expression of CD30 is a distinct feature of B- or T-cell activation, found in Hodgkin's disease, large cell anaplastic lymphoma, lymphomatoid papulosis, as well as in certain viral infections such as human T-lymphotropic virus type I, HIV, hepatitis B and C virus, and Epstein-Barr virus. Here, we report highly proliferative CD30-positive cutaneous infiltrates in 3 patients with Milkers's nodules, adding parapoxvirus infection to the spectrum of CD30-positive benign lympho-proliferations.
============================================================
28.) CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: A clue to the pathophysiology of clinical regression.
==========================================================
Blood 1999 Nov 1;94(9):3077-83
Mori M, Manuelli C, Pimpinelli N, Mavilia C, Maggi E, Santucci M, Bianchi B, Cappugi P, Giannotti B, Kadin ME Department of Dermatology, University of Florence Medical School, Florence, Italy.
Primary CD30(+) cutaneous T-cell lymphomas (CTCLs) represent a spectrum of non-Hodgkin's lymphomas (NHLs) that have been well defined at the clinical, histologic, and immunologic level. This group, which includes 2 main entities (large cell lymphoma and lymphomatoid papulosis [LyP]) and borderline cases, is characterized by the expression of CD30 antigen by neoplastic large cells at presentation, possible spontaneous regression of the skin lesions, and generally favorable clinical course.
Although the functional relevance of CD30 and its natural ligand (CD30L) expression in most cases of NHL is presently undefined, previous studies indicate that CD30L is likely to mediate reduction of proliferation in CD30(+) anaplastic large-cell NHL. No information is currently available concerning the expression of CD30L in primary CD30(+) CTCLs. In this study, we investigated the immunophenotypic and genotypic expression of CD30 and CD30L in different developmental phases of skin lesions (growing v spontaneously regressing).
By immunohistochemistry, CD30L expression was detected in regressing lesions only; by molecular analysis, the expression of CD30L was clearly higher in regressing lesions than in growing ones. CD30L, while expressed by some small lymphocytes, was most often coexpressed by CD30(+) neoplastic large cells, as demonstrated by 2-color immunofluorescence and by immunohistochemistry on paraffin sections. Taken together, these data suggest that CD30-CD30L interaction may play a role in the pathobiology of primary cutaneous CD30(+) lymphoproliferative disorders. In particular, CD30L (over)expression might have a major role in the mechanism of self-regression of skin lesions, the most distinctive clinical feature of this cutaneous lymphoma subtype.
============================================================
29.) Increased risk of lymphoid and nonlymphoid malignancies in patients with lymphomatoid papulosis.
============================================================
Cancer 1999 Oct 1;86(7):1240-5
Wang HH, Myers T, Lach LJ, Hsieh CC, Kadin ME Department of Pathology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts 02115, USA.
BACKGROUND: Lymphomatoid papulosis (LyP) is a rare skin disease with malignant potential. The long term outcomes of patients with this disease have not been adequately assessed.
METHODS: Fifty-seven patients with biopsy-proven LyP and 67 controls matched for age, gender, and race were followed prospectively from 1988 to 1996. Reported malignancies were confirmed by surgical pathology and/or autopsy reports. A search through the National Death Index through December 1995 was conducted to identify all deaths, and death certificates were procured. Expected numbers of malignancies based on SEER data were calculated for both the patient and the control groups.
RESULTS: Six LyP patients (10.5%) and 1 control (1.5%) reported nonlymphoid malignancies (P = 0.047). Two patients and no controls developed lymphoid malignancies (mycosis fungoides and CD30(+) cutaneous lymphoma). The expected numbers of nonlymphoid and lymphoid malignancies in the LyP patient group, based on the SEER data, were 1.93 and 0.15, respectively, yielding a relative risk (with 95% confidence interval) of 3.11 (1.26-6.47) for nonlymphoid malignancies and 13.33 (2.24-44.05) for malignant lymphomas in the LyP patients. There was no significant difference between the observed and expected numbers of malignancies in the control group. Four LyP patients died during the follow-up, three due to malignancies; and one control died of a gunshot wound to the head (suicide). The difference in overall survival between the LyP patients and the controls was not statistically significant (P = 0. 12).
CONCLUSIONS: Patients with LyP appear to have an increased risk of both lymphoid and nonlymphoid malignancies. The increased risk of nonlymphoid as well as lymphoid malignancies may suggest a basic underlying genetic defect leading to the development of malignancy in LyP patients. Copyright 1999 American Cancer Society.
============================================================
30.) Primary cutaneous CD8-positive epidermotropic cytotoxic T cell lymphomas. A distinct clinicopathological entity with an aggressive clinical behavior.
============================================================
Am J Pathol 1999 Aug;155(2):483-92
Berti E, Tomasini D, Vermeer MH, Meijer CJ, Alessi E, Willemze R Institute of Dermatologic Science, IRCCS Ospedale Maggiore, Milan, Italy. berti@jupiter.augustea.it
Cutaneous T cell lymphomas (CTCL) generally have the phenotype of CD3+, CD4+, CD45RO+ memory T cells. CTCL expressing a CD8+ T cell phenotype are extremely rare and ill-defined. To elucidate whether these CD8+ CTCL represent a distinct disease entity, the clinical, histological, and immunophenotypical features of 17 CD8+ CTCL were reviewed. None of the 17 cases expressed markers characteristic of natural killer cells or gamma/delta T cells. Nine of 17 cases showed the characteristic clinical and histological features as well as clinical behavior of well defined types of CTCL, such as mycosis fungoides (2 cases), pagetoid reticulosis (2 cases), lymphomatoid papulosis (2 cases), and CD30+ large T cell lymphoma (2 cases), all of which usually express a CD4+ T cell phenotype, and 1 case of subcutaneous panniculitis-like T cell lymphoma. The other 8 cases formed a homogeneous group showing a distinctive set of clinicopathological and immunophenotypical features, not consistent with that of other well defined types of CTCL.
Clinical characteristics included presentation with generalized patches, plaques, papulonodules, and tumors mimicking disseminated pagetoid reticulosis; metastatic spread to unusual sites, such as the lung, testis, central nervous system, and oral cavity, but not to the lymph nodes; and an aggressive course (median survival, 32 months).
Histologically, these lymphomas were characterized by band-like infiltrates consisting of pleomorphic T cells or immunoblasts, showing a diffuse infiltration of an acanthotic epidermis with variable degrees of spongiosis, intraepidermal blistering, and necrosis. The neoplastic cells showed a high Ki-67 proliferation index and expression of CD3, CD8, CD7, CD45RA, betaF1, and TIA-1 markers, whereas CD2 and CD5 were frequently lost. Expression of TIA-1 pointed out that these lymphomas are derived from a cytotoxic T cell subset.
The results of this and other studies reviewed herein suggest that these strongly epidermotropic primary cutaneous CD8+ cytotoxic T cell lymphomas represent a distinct type of CTCL with an aggressive clinical behavior.
============================================================
31.) Lymphomatoid papulosis in association with mycosis fungoides: a study of 15 cases.
============================================================
Br J Dermatol 1998 Oct;139(4):630-8
Basarab T, Fraser-Andrews EA, Orchard G, Whittaker S, Russel-Jones R St John's Institute of Dermatology, St Thomas' Hospital, London, UK.
We report clinical findings in 15 patients with lymphomatoid papulosis (LyP) associated with mycosis fungoides (MF). LyP either preceded (n = 4), followed (n = 5) or occurred concurrently with the MF lesions (n = 6). Twenty-eight LyP lesions were classified histologically and analysed further with immunostaining for CD3 and CD30.
Five biopsies contained a predominance of type A cells, six biopsies contained a predominance of type B cells. and six were mixed (A + B). However, 11 biopsies contained a population of atypical mononuclear cells with large hyperchromatic nuclei that we have termed indeterminate cells. These cells contained a thin rim of eosinophilic cytoplasm and showed strong CD30 but absent, faint or normal CD3 staining. In seven biopsies from five separate patients these cells represented the predominant cell type and we have termed this the pleomorphic variant of LyP. Analysis of T-cell receptor genes using Southern blot analysis and polymerase chain reaction/single strand conformational polymorphism analysis identified a T-cell clone in six of 16 LyP lesions and nine of 16 MF lesions. In the three patients who had clones in both types of skin lesions, the clones were identical.
Only two of 10 blood samples, both of which were from the same patient, had a T-cell clone and none of two lymph nodes showed evidence of a clonal population. To date all patients are alive with a median follow-up of 15 years from the onset of the first lesion. One patient has developed Lin anaplastic large cell lymphoma of the nasopharynx. These data augment the current literature on the association of LyP and MF and suggest that the
============================================================
32.) Early detection of cutaneous lymphoma.
============================================================
Oncology (Huntingt) 1998 Oct;12(10):1521-30; discussion 1532-4
Abd-el-Baki J, Stefanato CM, Koh HK, Demierre MF, Foss FM Department of Dermatology, Boston University School of Medicine, Massachusetts, USA.
Cutaneous lymphomas comprise a spectrum of diseases characterized by infiltration of the skin by malignant lymphocytes. The clinical manifestations of cutaneous lymphomas vary, and they can mimic benign dermatoses, as well as nodal or visceral malignancies with cutaneous spread.
Cutaneous lymphomas are divided into T-cell lymphomas and B-cell lymphomas. Cutaneous T-cell lymphomas include mycosis fungoides, Sezary syndrome, lymphomatoid papulosis, CD30+ large cell lymphoma, and adult T-cell leukemia/lymphoma. The extent and severity of skin manifestations in cutaneous T-cell lymphomas are prognostic indicators of extracutaneous involvement.
Primary cutaneous B-cell lymphomas comprise 10% to 25% of all primary cutaneous non-Hodgkin's lymphomas and are classified according to their cell of origin. Most cutaneous B-cell lymphomas have an indolent course and excellent prognosis when compared to their nodal counterparts. Many factors have been implicated in the etiology of cutaneous lymphomas, including chemical and drug exposures, as well as microbial agents, such as the Epstein-Barr virus (EBV), human T-lymphocyte virus-1 (HTLV-1), and Borrelia burgdorferi. Immunohistochemistry and lymphocyte-receptor gene rearrangement studies are useful in distinguishing malignant from benign conditions.
============================================================
33.) Cutaneous pseudolymphomas.
============================================================
J Am Acad Dermatol 1998 Jun;38(6 Pt 1):877-95; quiz 896-7
Ploysangam T, Breneman DL, Mutasim DF Department of Dermatology, University of Cincinna
ti Medical Center, Ohio, USA.
Cutaneous pseudolymphoma refers to a heterogeneous group of benign reactive T- or B-cell lymphoproliferative processes of diverse causes that simulate cutaneous lymphomas clinically and/or histologically. The inflammatory infiltrate is bandlike, nodular, or diffuse and is composed predominantly of lymphocytes with or without other inflammatory cells. Depending on the predominant cell type in the infiltrate, cutaneous pseudolymphomas are divided into T- and B-cell pseudolymphomas.
Cutaneous T-cell pseudolymphomas include idiopathic cutaneous T-cell pseudolymphoma, lymphomatoid drug reactions, lymphomatoid contact dermatitis, persistent nodular arthropod-bite reactions, nodular scabies, actinic reticuloid, and lymphomatoid papulosis. Cutaneous B-cell pseudolymphomas include idiopathic lymphocytoma cutis, borrelial lymphocytoma cutis, tattoo-induced lymphocytoma cutis, post-zoster scar lymphocytoma cutis, and some persistent nodular arthropod-bite reactions. This review attempts to discuss current aspects of the classification, pathogenesis, clinical spectrum, histopathologic and immunohistochemical diagnosis, and laboratory investigations for clonality in the various types of cutaneous pseudolymphomas.
============================================================
34.) Lymphomatoid papulosis in childhood with exclusive acral involvement.
============================================================
Pediatr Dermatol 1998 Mar-Apr;15(2):146-7
Thomas GJ, Conejo-Mir JS, Ruiz AP, Linares Barrios M, Navarrete M Publication Types:
Letter
============================================================
============================================================
35.) Absence of HTLV-1 proviral sequences in patients with lymphomatoid papulosis.
============================================================
J Invest Dermatol 1997 Dec;109(6):817-8
Ortiz Romero PL, Vallejo A, Lopez Estebaranz JL, Garcia Saiz A, Fernandez V, Iglesias Diez L Publication Types:
letter
============================================================
============================================================
36.) Clinicopathologic manifestations of Epstein-Barr virus-associated cutaneous lymphoproliferative disorders.
============================================================
Arch Dermatol 1997 Sep;133(9):1081-6
Iwatsuki K, Ohtsuka M, Harada H, Han G, Kaneko F Department of Dermatology, Fukushima Medical College, Japan.
OBJECTIVE: To elucidate clinicopathologic manifestations of cutaneous lymphoproliferative disorders associated with Epstein-Barr virus (EBV) infection. DESIGN: Retrospective survey of case series. SETTING: University hospital medical center. PATIENTS: Sixty-five patients with cutaneous lymphomas and related disorders.
MAIN OUTCOME MEASURES: Detection of EBV genes and EBV-encoded small nuclear RNAs.
RESULTS: Evidence of latent EBV infection was demonstrated in 15 patients: 3 had malignant lymphoma with clinical features mimicking cytophagic histiocytic panniculitis, 6 had facial vesiculopapular eruptions mimicking hydroa vacciniforme, 4 had angiocentric lymphoma, 1 had histiocytoid lymphoma associated with hemophagocytosis, and 1 had plasmacytoma. Hypersensitivity to mosquito bites was noted in a patient with hydroa vacciniforme-like eruptions and another with histiocytoid lymphoma.
Angiocentric infiltration of atypical lymphoid cells was a common histological feature in the patients with hydroa vacciniforme-like eruptions and angiocentric lymphoma. No evidence of EBV infection was apparent in 19 patients with mycosis fungoides or Sezary syndrome, 7 with adult T-cell leukemia or lymphoma, 3 with lymphomatoid papulosis (type A), and 2 with lymphocytoma cutis.
CONCLUSION: Patients with EBV-associated cutaneous lymphoproliferative disorders present with unique and diagnostic clinicopathologic features distinct from those of mycosis fungoides or Sezary syndrome.
============================================================
37.) Follicular lymphomatoid papulosis and multiple myeloma.
============================================================
Acta Derm Venereol 1997 Sep;77(5):403
Rongioletti F, Basso GI, Sementa A, Gambini C, Rebora A Publication Types:
Letter
============================================================ ============================================================
38.) Lymphomatoid papulosis--treatment with recombinant interferon alfa-2a and etretinate.
============================================================
SO - Dermatology 1995;190(4):288-91 AU - Wyss M; Dummer R; Dommann SN; Joller-Jemelka HI; Dours-Zimmermann MT; Gilliet F; Burg G PT -
JOURNAL ARTICLE
AB - Lymphomatoid papulosis is a rare cutaneous lymphoproliferative disorder with nodular, papulonecrotic or plaque-like lesions. Although it is clinically benign, the histology shows large, atypical lymphoid cells that display antigenic markers of activated T-helper lymphocytes and express CD30. There is a close relationship to Hodgkin's disease and to Ki-1-positive anaplastic large-cell lymphoma of the skin. For therapy, various modalities such as PUVA, steroids and acyclovir have been used. We report on a patient with a 10-year history of disease. Treatment with interferon alfa-2a, 3 MU 3 times/week for 4 weeks, and etretinate, 50 mg/day for 5 months, was initially successful, but lesions further relapsed 5 months after cessation of the therapy.
============================================================
39.) Lymphomatoid papulosis: a follow-up study of 41 patients.
============================================================
SO - Semin Dermatol 1994 Sep;13(3):197-201 AU - Christensen HK; Thomsen K; Vejlsgaard GL PT - JOURNAL ARTICLE
AB - Forty-one patients with lymphomatoid papulosis have been followed from 1 to 22 years (mean 11.4 years, median 10 years). Six patients developed malignant lymphoma, 3 cutaneous T-cell lymphoma, 2 Ki-1 large cell lymphoma, and 1 Hodgkin's disease. A clinical malignant presentation combined with the finding of aneuploidy in skin lesions seem to be indications of a malignant potential. Treatment with methotrexate in low dosage is an efficient treatment of lymphomatoid papulosis and probably diminishes the risk of malignancy.
============================================================
40.) Lymphomatoid papulosis associated with acquired ichthyosis.
============================================================
SO - J Am Acad Dermatol 1994 May;30(5 Pt 2):889-92 AU - Yokote R; Iwatsuki K; Hashizume H; Takigawa M PT -
JOURNAL ARTICLE
AB - We describe a 64-year-old man with lymphomatoid papulosis associated with acquired ichthyosis. The papulonodular lesions were composed of large atypical lymphocytes positive for CD3, CD4, and Ki-1. The ichthyosiform eruption also occurred on the extremities and had the histologic features of ichthyosis vulgaris. Although monoclonality of infiltrating cells could not be demonstrated, acquired ichthyosis appears to be induced in patients with lymphomatoid papulosis by the same pathomechanism underlying other lymphoproliferative diseases.
============================================================
41.) Primary anaplastic large-cell lymphoma of the skin. A case report suggesting that regressing atypical histiocytosis and lymphomatoid papulosis are subsets.
============================================================
SO - J Am Acad Dermatol 1994 Feb;30(2 Pt 2):358-63 AU - Yashiro N; Kitajima J; Kobayashi H; Fushida H; Nakagawa K; Furukawa M; Hamada T PT -
JOURNAL ARTICLE
AB - A patient with primary anaplastic large-cell lymphoma of the skin with characteristic clinical findings is described. The diagnosis was made on the basis of histologic and immunohistochemical findings. The phenotype of the tumor cells was not determined, but rearrangement of the T-cell receptor beta gene indicated that the tumor was of T-cell lineage. Despite high-grade malignancy of the tumor cells, the patient unexpectedly had a benign clinical course. The findings in this case suggest that regressing atypical histiocytosis and lymphomatoid papulosis type A are subsets of anaplastic large-cell lymphoma.
============================================================
42.) Lymphomatoid papulosis: a clinical and histopathologic review of 53 cases with leukocyte immunophenotyping, DNA flow cytometry, and T-cell receptor gene rearrangement studies.
============================================================
SO - J Am Acad Dermatol 1994 Feb;30(2 Pt 1):210-8 AU - el-Azhary RA; Gibson LE; Kurtin PJ; Pittelkow MR; Muller SA PT -
JOURNAL ARTICLE
AB - BACKGROUND: Lymphomatoid papulosis (LyP) is a recurrent hemorrhagic papular skin eruption with a clinically benign course and histopathologic features of lymphoma.
OBJECTIVE AND METHODS: To better characterize this disease, we studied 53 patients seen since 1965.
RESULTS: A lymphoproliferative malignancy developed within 2 to 36 years after onset of LyP in eight patients. Histologically, the dermis in LyP showed an infiltrate of large (type A) or small (type B) atypical lymphocytes. The large atypical cells (type A) stained with CD30 (Ber-H2). Seven of the patients in whom lymphoma developed had type A histologic features. DNA flow cytometry showed mainly a diploid pattern, except for two cases that showed aneuploidy. Five of 11 patients showed T-cell receptor (TCR) clonal gene rearrangements; lymphoma has not developed in these patients. One patient had a TCR rearrangement in a plaque of mycosis fungoides but not in the LyP lesion.
CONCLUSION: LyP is either a reactive skin condition or a localized lymphoid malignancy. Neither DNA flow cytometry nor TCR gene rearrangement can predict the 15% to 19% of patients in whom a lymphoma will develop. Continued observation of all patients is essential.
============================================================
43.) [Lymphomatoid papulosis and anaplastic giant-cell lymphoma]
============================================================
SO - Ann Dermatol Venereol 1994;121(10):727-30 AU - Moreau-Cabarrot A; Bonafe JL; Gorguet B; Andrieu H; Massip P PT -
JOURNAL ARTICLE
AB - INTRODUCTION.
The association between lymphomatoid papulosis and malignant Hodgkin or non-Hodgkin lymphoma is well known but still raises the problem of nosology between these two pathologies. Is lymphomatoid papulosis a pseudolymphoma, a prelymphomatous state or a true skin lymphoma? CASE REPORT.
We observed a patient who had lymphomatoid papulosis and anaplastic large-cell lymphoma within an interval of 8 years between. This case was particularly interesting because identical immunophenotypes were observed in the atypical large-cells of the skin and the lymphomatous cells of the lymph nodes (positive for CD43, CD45, CD25, CD30, CD15, EMA).
DISCUSSION. This case points out that atypical large-cells of lymphomatoid papulosis express the CD15 antigen which is only expressed by atypical large-cells in half of the cases of lymphomatoid papulosis. In addition, EMA is classically expressed in primary lymph node lymphomas rather than in primary cutaneous anaplastic large cell lymphomas which could predict extracutaneous dissemination of lymphomatoid papulosis. Furthermore, the demonstration that the skin lesions and the lymph nodes responded differently to the same treatment would suggest that there are other unrecognized biological differences. Lymphomatoid papulosis appears to be a range of disorders of the lymphoproliferation of activated T-cells and could include varioliform parapsoriasis and cutaneous lymphoma.
============================================================
44.) Molecular evidence for a clonal relationship between lymphomatoid papulosis and Ki-1 positive large cell anaplastic lymphoma.
============================================================
SO - J Dermatol Sci 1993 Oct;6(2):121-6 AU - Volkenandt M; Bertino JR; Shenoy BV; Koch OM; Kadin ME PT -
JOURNAL ARTICLE
AB - Currently, considerable controversy surrounds questions about the clonal evolution of lymphomas in patients with lymphomatoid papulosis. In order to analyze a possible clonal relationship between lesions of lymphomatoid papulosis and a Ki 1+ large cell anaplastic lymphoma in the same patient, a highly specific molecular probe for the malignant lymphoid clone of the large cell anaplastic lymphoma was developed. As a clone specific molecular marker, highly variable junctional sequences of rearranged T-cell receptor-gamma genes were used. An oligonucleotide primer complementary to these sequences was synthesized and, using the polymerase chain reaction, clone specific DNA was detected in all lesions of lymphomatoid papulosis of the patient. These results provide evidence for a clonal relationship between lesions of lymphomatoid papulosis and large cell anaplastic lymphoma developing in the same patient.
============================================================
45.) Lymphomatoid papulosis followed by Hodgkin's lymphoma. Differential response to therapy [see comments]
============================================================
SO - Arch Dermatol 1993 Jan;129(1):86-91 AU - Zackheim HS; Le Boit PE; Gordon BI; Glassberg AB PT - JOURNAL ARTICLE; REVIEW (32 references);
REVIEW OF REPORTED CASES
AB - BACKGROUND.
the association of lymphomatoid papulosis (LyP) with Hodgkin's lymphoma or other lymphomas is well recognized. However, the issue as to whether this represents an independent association or a transformation of one proliferative process to the other remains unresolved.
OBSERVATION. A woman with LyP subsequently developed Hodgkin's lymphoma. Combination chemotherapy resulted in apparent cure of the lymphoma but had only a transient effect on the LyP. A literature review revealed a similar difference in response to chemotherapy or radiation therapy in most patients who had LyP and associated lymphoma.
CONCLUSIONS. The differential response to therapy in patients with LyP and associated lymphoma suggests that there are biological differences between LyP cells and associated lymphoma cells even though in some patients the immunophenotype and genotype were reported to be identical. However, alternative explanations are possible. In this article we also review studies on other cases of LyP associated with Hodgkin's lymphoma.
============================================================
46.) Epidemiology of lymphomatoid papulosis [see comments]
============================================================
SO - Cancer 1992 Dec 15;70(12):2951-7 AU - Wang HH; Lach L; Kadin ME PT
- JOURNAL ARTICLE
AB - BACKGROUND.
Lymphomatoid papulosis is a rare skin disease with malignant potential. Its epidemiology is largely unknown.
METHODS. A case-control study of lymphomatoid papulosis was done to characterize the patient population and investigate the risk factors for its development. Fifty-seven patients with biopsy-proven lymphomatoid papulosis and 67 individually matched control subjects who were recruited among relatives and acquaintances of the patients answered a standard questionnaire over the telephone.
RESULTS. Among patients with lymphomatoid papulosis, 3 had a history of Hodgkin disease, 3 had non-Hodgkin lymphoma, and 10 had mycosis fungoides; none of the control subjects reported such histories. No significant differences were observed between patients and control subjects in regard to residence or travel history or exposures to various physical, chemical, and biologic agents. A higher, although not statistically significant, percentage of patients than control subjects reported a history of radiation therapy and nonlymphoid malignant lesions. No differences were found between patients and control subjects in regard to other medical conditions or family medical history.
CONCLUSIONS. Patients with lymphomatoid papulosis have a significantly increased frequency of prior or coexisting lymphoproliferative disorders, an increased frequency of nonlymphoid malignant lesions, and exposure to radiation therapy.
============================================================
47.) Lethal midline granuloma (peripheral T-cell lymphoma) after lymphomatoid papulosis.
============================================================
SO - Cancer 1992 Aug 15;70(4):835-9 AU - Harabuchi Y; Kataura A; Kobayashi K; Yamamoto T; Yamanaka N; Hirao M; Onodera K; Kon S PT -
JOURNAL ARTICLE
AB - A Japanese woman with an 8-year history of lymphomatoid papulosis (LP) had lethal midline granuloma (LMG) develop at the age of 51 years. There were histologic similarities between LP and LMG seen in this patient. Surface phenotypic studies on nasal and cutaneous lesions demonstrated a population of T-cells expressing CD2, CD4, CD25, CD30, and histocompatibility antigen-DR (HLA-DR). Genotypic analyses of nasal and skin biopsy specimens disclosed a clonal rearrangement of the beta T-cell receptor gene with the same rearrangement pattern. These data indicate that this patient had LMG characterized by clonal peripheral T-cell lymphoma, which probably resulted from progression of the LP.
============================================================
48.) The prognosis of patients with lymphomatoid papulosis associated with malignant lymphomas.
============================================================
SO - Br J Dermatol 1992 Jun;126(6):596-602 AU - Beljaards RC; Willemze R PT - JOURNAL ARTICLE; REVIEW (38 references);
REVIEW OF REPORTED CASES
AB - Lymphomatoid papulosis (LyP) is a disorder which generally runs a benign course, but can sometimes be associated with a malignant lymphoma. Information about the prognosis of these LyP-associated lymphomas is, however, fragmentary. In this study, the clinical data of 50 LyP-associated malignant lymphomas, including 11 patients of our own group and 39 reported in the literature, are evaluated. Three main groups of LyP-associated malignant lymphomas could be distinguished: cases associated with mycosis fungoides (19/50 cases). Hodgkin's disease (12/50 cases) and (CD30+) large-cell lymphomas (16/50). The results of this study demonstrate that patients with mycosis fungoides. Hodgkin's disease, and (CD30+) large-cell lymphomas limited to the skin have a favourable prognosis. However, the prognosis of patients developing a systemic (CD30+) large-cell lymphoma proved generally poor. The results of this study also indicate that the risk of an individual LyP patient developing systemic lymphoma is less than 5%.
============================================================
49.) Immunophenotypic and genotypic characterization of lymphomatoid papulosis.
============================================================
SO - J Am Acad Dermatol 1992 Jun;26(6):968-75 AU - Parks JD; Synovec MS; Masih AS; Braddock SW; Nakamine H; Sanger WG; Harrington DS; Weisenburger DD PT - JOURNAL ARTICLE; REVIEW (24 references);
REVIEW, TUTORIAL AB -
BACKGROUND: Lymphomatoid papulosis (LyP) is a chronic dermatosis that histologically resembles malignant lymphoma. Thus far, only a few cases of LyP have been characterized in detail with regard to immunophenotype, genotype, and karyotype.
OBJECTIVE: Our purpose was to study seven patients with LyP and compare the results to those reported in the literature.
METHODS: Skin biopsy specimens were analyzed by frozen section immunohistochemical and molecular biologic techniques. Cytogenetic analysis was also performed in three cases.
RESULTS: The atypical lymphoid cells consisted of activated helper T cells; four of the seven patients had lesions with a detectable clonal T-cell population. A peripheral T-cell lymphoma developed in one patient before the emergence of a genotypically different LyP T-cell clone. Cytogenetic studies were abnormal in one case of LyP and normal in another, whereas the karyotype of the lymphoma was abnormal.
CONCLUSION: LyP is a preneoplastic proliferation of activated helper T cells, which is often clonal and may regress and expand with the development of new LyP clones or lymphoma.
============================================================
50.) Pityriasis lichenoides and lymphomatoid papulosis.
============================================================
SO - Semin Dermatol 1992 Mar;11(1):73-9 AU - Rogers M PT -
JOURNAL ARTICLE; REVIEW (79 references); REVIEW, TUTORIAL
AB - The clinical features, histopathology, immunopathology, and management of pityriasis lichenoides and lymphomatoid papulosis are discussed, with particular emphasis on the pediatric aspects of these conditions. The difficulties in logically separating pityriasis lichenoides into an acute (pityriasis lichenoides et varioliformis acuta) and a chronic (pityriasis lichenoides chronical) form are addressed.
The development of lymphoreticular malignancy in patients with lymphomatoid papulosis has been well documented, but pityriasis lichenoides has characteristically been regarded as a benign condition. However, recent reports of the development of large plaque parapsoriasis in patients with pityriasis lichenoides have led to a reconsideration. Some of these patients were in the pediatric age group.
Although there are significant clinical, histopathological, and immunopathological differences between pityriasis lichenoides and lymphomatoid papulosis, the demonstration of similar clonal T cell receptor gene rearrangements and the confirmation of the potentially premalignant nature of both suggests that there may indeed be an interrelationship between these two controversial entities. Close follow-up of patients with both of these conditions is recommended, with observation being discontinued only when the patient has been free of lesions for several years.
============================================================
51.) Lymphomatoid papulosis: clinicopathological comparative study with pityriasis lichenoides et varioliformis acuta.
============================================================
SO - J Dermatol 1991 Oct;18(10):580-5 AU - Erpaiboon P; Mihara I; Niimura M PT -
JOURNAL ARTICLE
AB - We have compared the clinical and histopathological features of 6 patients with lymphomatoid papulosis (LP) and 14 patients with pityriasis lichenoides et varioliformis acuta (PLEVA). There were some differences between the clinical features in the two diseases, including the size and appearance of skin lesions and the duration of the course of disease. Ki-1 Ag positive, large, atypical, lymphoid cells were always seen in lymphomatoid papulosis; none of lymphoid cells of pityriasis lichenoides et varioliformis acuta demonstrated this antigen. We conclude that lymphomatoid papulosis and PLEVA, although sharing some common features, should be considered to be different clinical and immunopathological entities.
============================================================
52.) PUVA-induced lymphomatoid papulosis in a patient with mycosis fungoides.
============================================================
SO - J Am Acad Dermatol 1991 Aug;25(2 Pt 2):422-6 AU - Wolf P; Cerroni L; Smolle J; Kerl H PT - JOURNAL ARTICLE
AB -
The occurrence of lymphomatoid papulosis in patients with cutaneous lymphoma, particularly mycosis fungoides, has been described in medical literature. A 68-year-old woman affected by mycosis fungoides in the plaque stage noticed that multiple papulonodular lesions of lymphomatoid papulosis developed suddenly after a few sessions of PUVA therapy. The PUVA induction of lymphomatoid papulosis was confirmed by the appearance of new lesions after a second cycle of PUVA exposure on a limited area of the body. Complete regression of all PUVA-induced lymphomatoid papulosis lesions was achieved within a few weeks with oral prednisone and topical steroids. During the entire treatment the patches and plaques of mycosis fungoides persisted unchanged.
============================================================
53.) Accuracy in diagnosis of lymphomatoid papulosis.
============================================================
SO - Am J Dermatopathol 1991 Feb;13(1):20-5 AU - Cockerell CJ; Stetler LD PT -
JOURNAL ARTICLE
AB - This study was undertaken to assess the accuracy of histologic diagnosis of lymphomatoid papulosis (LyP), which may be confused with malignant lymphoma or other entities. It is essential that accurate diagnoses be made because LyP may be a marker for malignant lymphoma. All 15 examples of LyP reviewed in a dermatopathology laboratory during a 14-year period and 180 histologic sections of tissue that could be confused with LyP were reviewed. Criteria for diagnosis of LyP were applied without benefit of clinical history, and revised diagnoses were made where indicated.
Clinical follow-up information was obtained and original accuracy of diagnosis was assessed by comparing clinical courses with original histologic diagnoses. In cases of LyP in which numerous atypical lymphoid cells were present, 100% accuracy was noted. When fewer atypical lymphoid cells were present and inflammatory cell infiltrates were less dense, the diagnosis was less certain.
Overall, a 64% correlation of clinical course and histologic diagnosis of LyP was noted. We conclude that the histologic diagnosis of LyP is generally reliable and accurate; however, in some cases a precise diagnosis cannot be made with certainty. Cases with fewer atypical lymphoid cells may fail to correlate well with the classic course of LyP and may represent a variant or histologic simulator.
============================================================
54.)Regressing atypical histiocytosis and lymphomatoid papulosis: variants of the same disorder?
============================================================
SO - Br J Dermatol 1990 Oct;123(4):515-21 AU - Cerio R; Black MM PT -
JOURNAL ARTICLE
AB - We report a patient with lymphomatoid papulosis who developed a lesion with the clinicopathological features of regressing atypical histiocytosis. Immunohistochemical studies supported a T-cell histogenesis and many of the atypical cells demonstrated BerH2 (Ki-I antigen) positivity. The case supports the view that regressing atypical histiocytosis and lymphomatoid papulosis are different manifestations of the same disease spectrum.
============================================================
55.) Lymphomatoid papulosis and its relationship to "idiopathic" hypereosinophilic syndrome.
============================================================
SO - J Am Acad Dermatol 1988 Feb;18(2 Pt 1):339-44 AU - Whittaker SJ; Jones RR; Spry CJ PT - JOURNAL ARTICLE
AB - Persistent hypereosinophilia, endomyocardial fibrosis, and a recurrent self-healing papulonodular eruption with the histologic features of lymphomatoid papulosis are described in three patients. One patient died after developing an acute myeloblastic transformation in the eosinophil series. Immunocytochemical studies of cutaneous lesions in two of the patients suggested a mature T-cell phenotype with a predominant population of CD4-positive cells. Immunostaining of cutaneous tissue with monoclonal antibodies BE1 and BE2 yielded negative findings. Because it is now known from in vitro studies that T lymphocytes secrete the eosinopoietic factor, interleukin 5, it is possible that the cutaneous lesions, hypereosinophilia, and associated endomyocardial fibrosis were induced by transformed helper T lymphocytes in these three patients.
============================================================
56.) Lymphomatoid papulosis/pityriasis lichenoides in two children.
============================================================
SO - Pediatr Dermatol 1987 Nov;4(3):238-41 AU - Ashworth J; Paterson WD; MacKie RM PT - JOURNAL ARTICLE
AB - Two children developed lymphomatoid papulosis/pityriasis lichenoides at ages 3 and 6 years. Follow-up continued for 13 years in the former patient and for 6 years in the latter. Both children now have continuing low-grade disease activity requiring in the one case topical corticosteroid therapy and in the other low-dose systemic steroid therapy. These children are reported to emphasize to pediatricians, pediatric pathologists, and hematologists that pseudolymphomatous conditions can exist in young children and do not require potent cytotoxic therapy. In both of our patients, the initial diagnosis was thought to be an aggressive lymphoma.
============================================================
57.) [Acyclovir in mycosis fungoides and lymphomatoid papulosis]
============================================================
SO - Hautarzt 1986 Oct;37(10):533-6 AU - Burg G; Klepzig K; Kaudewitz P; Wolff H; Braun-Falco O PT - CLINICAL TRIAL;
JOURNAL ARTICLE
AB - A survey is given on 23 patients (10 of our own, 13 reported in personal communications and in the literature) suffering from lymphoproliferative diseases and treated with acyclovir (ACV). In 5 patients (3 of 18 with cutaneous T-cell lymphomas, 2 of 5 with lymphomatoid papulosis) partial remission could be achieved. Since herpes simplex virus, cytomegalovirus and viruses like Epstein-Barr and varicella-zoster do not play an etiologic role and since HTLV-I virus, due to its lack of thymidine kinase, cannot activate ACV, the following mechanisms should be discussed regarding the possible effectiveness of ACV in lymphoproliferative diseases: a direct cytopathic effect; activation of ACV by the thymidine kinase of viruses not yet detected in cutaneous lymphoproliferative disorders; ACV activation by cellular thymidine kinase, which has been found to be elevated in lymphoproliferative disorders. Preliminary clinical observations suggest that ACV may exhibit an antiproliferative effect intravenously in some patients with lymphomatoid papulosis.
============================================================
58.) Eosinophilic histiocytosis: a variant form of lymphomatoid papulosis or a disease sui generis?
============================================================
SO - J Am Acad Dermatol 1985 Dec;13(6):952-8 AU - McLeod WA; Winkelmann RK PT - JOURNAL ARTICLE
AB - Five patients are reported on whose clinical skin disease consisted of polymorphous papulonodular lesions healing with a depigmented scar. Although all cases had been termed lymphomatoid papulosis after clinical or histologic examination, the lesions consisted principally of masses of histiocytes and eosinophils. Individual lesions healed spontaneously or with minimal treatment, but the chronic course of disease was not altered by any therapy used. Follow-up 3 to 17 years later indicated persistent or recurrent disease, and one patient died of histiocytic malignancy. Eosinophilic histiocytosis is the microscopic picture of an unusual group of patients with chronic papulonodular necrotic skin disease that may deserve to be considered a disease pattern per se.
============================================================
59.) Lymphomatoid papulosis: a premalignant T cell disorder.
============================================================
SO - J Am Acad Dermatol 1985 Nov;13(5 Pt 1):736-43 AU - Espinoza CG; Erkman-Balis B; Fenske NA PT - JOURNAL ARTICLE
AB - In an attempt to better define the process of lymphomatoid papulosis, two cases were studied by means of light and electron microscopy, immunohistochemistry studies, including the use of monoclonal antibodies, and cytogenetic technics. About 90% of the dermal lymphoid infiltrate, including the atypical cells, reacted with antibodies that define helper-inducer T cells. Only a few cells, about 5%, reacted with antibodies that define cytotoxic-suppressor T cells. Langerhans cells were increased mostly within the epidermis, and in the dermis they were in close proximity to lymphoid cells. Cytogenetic studies disclosed an abnormal hypertetraploid karyotype in dividing cells from the skin lesion, whereas skin fibroblast and phytohemagglutinin-stimulated cells from peripheral blood cultures had a diploid karyotype. The results support the concept that lymphomatoid papulosis is a disorder characterized by a predominance of helper-inducer T cells, including the atypical cells bearing an abnormal karyotype.
============================================================
60.) Clinical and histologic differentiation between lymphomatoid papulosis and pityriasis lichenoides.
============================================================
SO - J Am Acad Dermatol 1985 Sep;13(3):418-28 AU - Willemze R; Scheffer E PT -
JOURNAL ARTICLE
AB - The relationship between lymphomatoid papulosis and pityriasis lichenoides is a matter of considerable debate. Differentiation between these two conditions is, however, important because patients with lymphomatoid papulosis, unlike those with pityriasis lichenoides, may develop systemic lymphoma and thus require long-term follow-up. In our study the clinical and histologic features of eighty-two patients with pityriasis lichenoides and twenty-six patients with lymphomatoid papulosis were reviewed and compared. Clinical and histologic differences were recognized, not only allowing differentiation between the two conditions, but also suggesting that they are pathogenetically distinct diseases. Finally, evidence is presented to suggest that the different views on the relationship between these diseases mainly result from differences in patient selection.
============================================================
61.) The clinicopathologic spectrum of lymphomatoid papulosis: study of 31 cases.
============================================================
SO - J Am Acad Dermatol 1983 Jan;8(1):81-94 AU - Sanchez NP; Pittelkow MR; Muller SA; Banks PM; Winkelmann RK PT -
JOURNAL ARTICLE
AB - Herein we review the Mayo Clinic experience with thirty-one cases of lymphomatoid papulosis seen since 1965. All patients had chronic, recurrent, and self-healing erythematous papulonodular lesions, which often became pustular, ulcerated, and resolved with scarring. The clinical features often corresponded to those seen in Mucha-Habermann disease; however, the predominant histopathologic feature was an infiltrate composed primarily of atypical lymphoid cells suggestive of malignant lymphoma. In six patients, a lymphoproliferative disorder was eventually diagnosed. There were two cases of mycosis fungoides (stage I), one case of nodular sclerosing Hodgkin's disease, and three cases of malignant lymphoma--one diffuse mixed large and small cell type with features of T-immunoblastic type, one diffuse large cell type, and one follicular small cleaved cell type. The clinical course of the lymphomatoid papulosis was unaffected by chemotherapy for the lymphoproliferative disorder. Our data indicate that, with sufficient duration of follow-up, malignant lymphoma may develop in some patients with lymphomatoid papulosis.
============================================================
62.) [Lymphomatoid papulosis in a child]
============================================================
SO - Hautarzt 1993 Oct;44(10):674-9 AU - Milde P; Goerz G; Lehmann P PT - JOURNAL ARTICLE; REVIEW (25 references);
REVIEW OF REPORTED CASES
AB - We report a case of a 3-year-old boy who developed crops of papules and ulcerating nodules on the limbs in April 1992. Periodically, new lesions continue to erupt, while others resolve spontaneously. This course is characteristic for rhythmic paradoxical eruptions. This course and the clinical picture, supported by the histopathological and immunohistochemical findings, led to the diagnosis of lymphomatoid papulosis. Lymphomatoid papulosis is extremely rare in childhood. All published cases of lymphomatoid papulosis in children under 10 years of age are reviewed. The differential diagnosis of lymphomatoid papulosis in childhood includes arthropod assaults, pityriasis lichenoides et varioliformis acuta, primary cutaneous Hodgkin's disease, Ki-1 large cell anaplastic lymphoma and other lymphomas and pseudolymphomas in children.
============================================================
63.)Lymphomatoid papulosis in an 11-month-old infant.
============================================================
SO - Pediatr Dermatol 1984 Oct;2(2):124-30 AU - Rogers M; de Launey J; Kemp A; Bishop A PT - JOURNAL ARTICLE
AB - Lymphomatoid papulosis was seen in an 11-month-old child. The condition resolved spontaneously after a course of only 8 weeks and the patient has now been disease free for 9 months. Electron microscopy showed infiltrating lymphocytes with cleaved nuclei suggestive of T cells. Monoclonal antibody studies confirmed the T cell nature of the infiltrate. In this case, suppressor (OKT8) T cells were more prominent than helper (OKT4) T cells, in contrast to previous reports.
============================================================
64.) Topical carmustine therapy for lymphomatoid papulosis.
============================================================
SO - Arch Dermatol 1985 Nov;121(11):1410-4 AU - Zackheim HS; Epstein EH Jr; Crain WR PT - JOURNAL ARTICLE
AB - Seven patients with lymphomatoid papulosis were treated with solutions of topical carmustine, a nitrosourea compound. Recently used schedules have employed 10 mg of carmustine in dilute alcohol applied to the total skin surface daily for four to 17 weeks (total dosage, 280 to 1,180 mg). All patients experienced a rapid reduction in the number and size of lesions. Maintenance therapy consisted of local applications of carmustine (2 to 4 mg/mL of 95% ethanol) to individual new papules. This method was effective in suppressing disease activity and reduced by half the average life cycle of individual lesions. However, long-term lesion-free remissions were not seen. Bone marrow depression did not occur.
============================================================
65.) Immunohistology of pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica. Evidence for their interrelationship with lymphomatoid papulosis.
============================================================
SO - J Am Acad Dermatol 1987 Mar;16(3 Pt 1):559-70 AU - Wood GS; Strickler JG; Abel EA; Deneau DG; Warnke RA PT -
JOURNAL ARTICLE
AB - Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica are idiopathic, papular eruptions that exhibit certain clinicopathologic similarities to each other and to lymphomatoid papulosis. In order to determine if these disorders are also similar immunologically, we studied the immunopathology of five biopsy specimens from three cases of pityriasis lichenoides et varioliformis acuta and three biopsy specimens from three cases of pityriasis lichenoides chronica. We then compared them to our prior immunohistologic study of nine cases of lymphomatoid papulosis. Pityriasis lichenoides et varioliformis acuta and pityriasis lichenoides chronica both exhibited a dermal and epidermal infiltrate of CD4+ and CD8+ T cells expressing activation antigens. These were admixed with numerous macrophages.
The lesional epidermis was diffusely human lymphocyte antigen (HLA)-DR+ and contained decreased CD1+ dendritic cells. Endothelial cells were also HLA-DR+. Cells bearing the phenotypes of B cells, follicular dendritic cells, or natural killer/killer cells were essentially absent. Except for the lack of large atypical cells, the results resembled those described previously for lymphomatoid papulosis. These findings indicate that pityriasis lichenoides chronica, pityriasis lichenoides et varioliformis acuta, and lymphomatoid papulosis share several immunohistologic features. Together with certain clinicopathologic similarities, they are consistent with the hypothesis that these three disorders are interrelated.
===================================================================
DATA-MÉDICOS/DERMAGIC-EXPRESS No 2-(86) 12/01/2000 DR. JOSÉ LAPENTA R.
UPDATED 09 JULY 2025
===================================================================
Produced by Dr. José Lapenta R. Dermatologist
Venezuela 1.998-2.025
Producido por Dr. José Lapenta R. Dermatólogo Venezuela 1.998-2.025
Tlf: 0414-2976087 - 04127766810












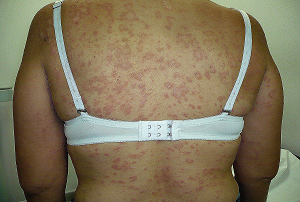




.png)