
LA ESCLEROSIS SISTÉMICA, ESCLERODERMIA
")
La esclerodermia, también conocida como esclerosis sistémica (ESc) es una enfermedad clasificada dentro de las colagenosis, es una enfermedad autoinmune crónica que se caracteriza por el engrosamiento y endurecimiento de la piel y también en algunos casos de los órganos internos, ocasionado esto por un aumento en la síntesis del colágeno, lo que lleva a la fibrosis de la piel y órganos internos (no todos los casos).
Este proceso es impulsado por fibroblastos y miofibroblastos activados, por factores inmunológicos que producen un aumento de la matriz extracelular, conduciendo a los síntomas descritos.
El nombre Esclerodermia viene del griego donde "esclero" significa duro y "derma", piel, de modo que la GRAN CARACTERÍSTICA DISTINTIVA de esta enfermedad es el endurecimiento de la piel, la cual al simple tacto se nota endurecida, a veces definida como "piel de cartón" o "acartonamiento" de la piel, obviamente más otros síntomas clínicos y hallazgos de laboratorio que tipifican esta patología.
ETIOLOGÍA:
La esclerosis sistémica tiene como desencadenantes factores tipo predisposición genética, y ambientales.
1.) Factores ambientales:
Entre los factores ambientales desencadenantes de esclerodermia esta de primera linea los trabajadores de las MINAS DE SÍLICE, los cuales al inhalar el polvo no solo desarrollan fibrosis pulmonar, sino que al tiempo desarrollan manifestaciones cutáneas típicas de la esclerosis sistémica. hecho que ha sido comprobado en diversos estudios (Canadá, China y Australia).
2.) Predisposición genética:
Etnias: con respecto a esto se ha descrito o descubierto que la población Afroamericana, tiende a padecer con mayor frecuencia de esta enfermedad en relación con los Europeos, tambien un inicio mas temprano de la enfermedad en la cual predomina el tipo difuso y mayor asociación con los anticuerpos ANTI-topoisomerasa I (SCL-70). Los Europeos presentan una mayor asociación con los anticuerpos ANTI-centrómero y una forma más leve de la enfermedad.
Antígenos HLA: Bien descrito en la literatura la asociación de varios ALELOS del Sistema Mayor de Histocompatibilidad con esta enfermedad, estos son: HLA-DRB111, HLA-DRB111:04, HLA-DPB1, HLA-DPB113:01, HLA-DPB113:01, HLA-DQA1 y HLA-DQB1, HLA-DQA105:01.
De este tema, hablaremos en nuestra próxima revisión titulada HLA Y ENFERMEDADES DEL TEJIDO CONECTIVO.
SÍNTOMAS:
Los síntomas de la esclerosis sistémica (ESc) clásica se dividen en:
1.) MANIFESTACIONES CUTÁNEAS:
A.- Endurecimiento de la piel: (fibrosis), particularmente de los dedos (esclerodactilia) y cara, en donde al producirse el endurecimiento de la piel la cara adopta un aspecto descrito como "cara de pajarito", poco descrito hoy dia. Otras manifestaciones cutáneas incluyen: telangiectasias periungueales (típicas de la esclerosis sistémica) , calcinosis , poikilo dermatosis, y úlceras digitales.
B.- Daño vascular: el daño en las células endoteliales de los vasos sanguíneos afecta dedos de manos y pies, produciéndose el FENÓMENO DE RAYNAUD (CLICK) que se caracteriza por cambios de coloración en la piel por efecto del frío, el cual que provoca vasoespasmo.
2.) MANIFESTACIONES SISTÉMICAS:
La fibrosis o endurecimiento de los tejidos orgánicos internos producen principalmente:
A.-Afectación pulmonar: hipertensión arterial pulmonar y enfermedad pulmonar intersticial (neumonitis intersticial).
B.- Afectación renal: caracterizada por insuficiencia renal aguda e hipertensión la cual puede ser grave.
C.- Afectación cardíaca: la fibrosis del tejido cardiaco puede provocar alteraciones miocárdicas, arritmias, y derrame pericárdico.
D.-Afectación gastrointestinal: el endurecimiento del tejido gastrointestinal puede provocar: disminución de la motilidad esofágica, produciendo dificultad para deglutir alimentos, reflujo gastroesofágico y pseudo obstrucción intestinal.
E.- Afectación de articulaciones y músculos: artralgias y mialgias.
F.-Malestar general: fatiga.
CLASIFICACIÓN:
Algunas escuelas dermatológicas hablan o clasifican la esclerodermia en:
1.) ESCLERODERMIA LOCALIZADA: que viene siendo la misma
A.) Esclerodermia Localizada o Morfea (CLICK), donde no HAY SÍNTOMAS SISTÉMICOS; en el tema MORFEA están bien explicados los términos de MORFEA, y ESCLERODERMIA LOCALIZADA LINEAL.
B.) Esclerodermia en "Cup de Sabre": es una variante de la esclerodermia localizada que se manifiesta SOLO en la región frontal, en forma de hundimiento o atrofia lineal que llega hasta el cuero cabelludo, dando el aspecto de un "hachazo" o "hendidura de sable". Algunos la consideran una variante de la llamada atrofia hemifacial progresiva. No se han encontrado anticuerpos específicos de esclerodermia, ni hay síntomas sistémicos en la mayoría de los casos.
2.) ESCLEROSIS SISTÉMICA (ESc): la cual se subdivide en tres grupos hoy dia 2024.
A.- Esclerosis sistémica cutánea difusa (EScd):
Es la de peor pronóstico, pues la fibrosis y afectación de la piel es extensa, prácticamente incluyendo casi todo el cuerpo, principalmente torso y brazos. La fibrosis además afecta órganos internos (corazón, riñón, pulmones) con una progresión rápida en etapas tempranas de la enfermedad y por lo tanto es la peor de todas en cuanto a tiempo de sobrevida.
B.-Esclerosis sistémica cutánea limitada (EScl):
Este subtipo era conocido y descrito como SÍNDROME DE CREST, una variante de esclerodermia limitada donde la fibrosis afecta fundamentalmente cara y brazos, sus características son:
C: calcinosis.
R: Fenómeno de Raynaud.
E: Esclerodactilia.
S: Trastornos de la motilidad esofágica.
T: Telangiectasias periungueales. (típico)
Esta variante representa un 20% de los casos de esclerodermia, tiene mejor pronóstico, su curso es más lento y está asociada principalmente a anticuerpos ANTI- centrómero.
Estas eran las dos clasificaciones clásicas de la esclerosis sistémica, pero posteriormente se describió al igual que en la DERMATOMIOSITIS (CLICK), sin miositis, una variante denominada:
C.-Esclerosis sistémica sine esclerodermia (ESss):
En esta variante NO HAY FIBROSIS CUTÁNEA, pero hay afectación de uno o mas órganos internos y representa el 10% de los casos de esclerodermia. Este subtipo puede se diagnosticado erróneamente porque no tiene manifestaciones cutáneas, pero a pesar de la ausencia de fibrosis, pueden haber manifestaciones graves cardiacas, renales (crisis renal esclerodérmica) y pulmonares (hipertensión arterial).
Para que esta variante sea diagnosticada, además de los síntomas descritos, en los hallazgos de laboratorio deben encontrarse algunos de los anticuerpos típicos de la esclerodermia, a saber:
Anticuerpos antinucleares (ANA), Anticuerpos anticentrómero (ACA), Anticuerpos anti-RNA polimerasa III, o Anticuerpos anti-Scl-70 (topoisomerasa I) y otros.
DIAGNÓSTICO:
En el año 2015 el Colegio Americano de Reumatología (ACR) y la Liga Europea contra el Reumatismo (EULAR) establecieron los siguientes criterios para el diagnóstico de esclerodermia:
1.) Engrosamiento de la piel de los dedos hasta las articulaciones metacarpofalángicas y / o lesiones en la punta de los dedos.
2.) Telangiectasias periungueales (capilares anormales en el pliegue ungueal).
3.) Afectación pulmonar.
4.) Fenómeno de Raynaud.
5.) Autoanticuerpos específicos para esclerodermia:
A.-Anticuerpos antinucleares (ANA): Presentes en más del 90% de los pacientes con esclerosis sistémica (ESc). Su ausencia no excluye el diagnóstico. Los ANA son predictores independientes de daño microvascular digital.
B.-Anticuerpos anti-topoisomerasa I (ATA-SCL-70): Asociados con la esclerosis sistémica cutánea difusa (EScd), úlceras en punta de los dedos, y enfermedad pulmonar severa (neumonitis intersticial).
C.-Anticuerpos anticentrómero (ACA): Asociados con la esclerosis sistémica cutánea limitada (EScl) y menor fibrosis pulmonar y daño renal, (el antiguo SÍNDROME DE CREST).
D.-Anticuerpos anti-RNA polimerasa III (ARA): Asociados con progresión rápida del engrosamiento cutáneo, crisis renal y asociación con cáncer.
E.-Anticuerpos anti-U3 RNP: Relacionados con hipertensión pulmonar, y complicaciones gastrointestinales (pacientes de etnia Afro Caribe).
G.-Anticuerpos anti-PM/Scl: Asociados a alta frecuencia de enfermedad pulmonar intersticial y miopatía.
H.-Anticuerpos anti-Ku: Asociados con síndrome de OVERLAP polimiositis/esclerosis sistémica, sin manifestaciones vasculares prominentes.
RESUMIENDO. HOY DIA 2024, LA CLASIFICACIÓN DE LA ESCLEROSIS SISTÉMICA SON LAS TRES (3) PREVIAMENTE DESCRITAS.
1.) CUTÁNEA DIFUSA O GENERALIZADA. (EScd)
2.) CUTÁNEA LOCALIZADA O LIMITADA (EScl), anterior síndrome de CREST.
3.) SINE ESCLERODERMIA (ESss), sin fibrosis cutánea.
TRATAMIENTO:
La esclerosis sistémica NO TIENE CURA, los tratamientos están dirigidos a mejorar los síntomas específicos de cada presentación clínica y son:
1.) MANEJO DE SÍNTOMAS ESPECÍFICOS:
A.-Fenómeno de Raynaud: nifedipina (antagonistas del calcio) e inhibidores de la fosfodiesterasa tipo 5 (PDE-5), sildenafil y tadalafil para mejorar la circulación y prevenir o reducir la frecuencia de los ataques.
-Iloprost intravenoso: puede utilizarse para ataques severos o úlceras digitales.
B.-Afectación Pulmonar:
- Ciclofosfamida: Utilizada en casos severos de esclerodermia difusa con afectación pulmonar, aunque su uso a largo plazo está limitado por su toxicidad.
- Micofenolato de mofetilo: al igual que la ciclofosfamida es utilizado en la esclerosis sistémica severa con enfermedad pulmonar intersticial.
- Pirfenidona: Aunque no está aprobada específicamente para la esclerosis sistémica con afectación pulmonar, se está investigando su uso en combinación con micofenolato de mofetilo.
- Bosentan, epoprostenol y otros análogos de prostaciclina: para el tratamiento de la hipertensión pulmonar y también puede reducir el desarrollo de nuevas úlceras digitales
C.-Afectación Renal:
- Enalapril, lisinopril: Los inhibidores de la enzima convertidora de angiotensina (IECA) son los más recomendados para tratar crisis renal esclerodérmica, mejorando significativamente la supervivencia.
- Losartán, valsartán: Antagonistas de los Receptores de Angiotensina II (ARA-II): Se utilizan como alternativa a los IECA. Ayudan a controlar la presión arterial y pueden mejorar el manejo de la función renal.
D.- Afectación Digestiva:
- Omeprazol, esomeprazol, lansoprazol, Pantoprazol, y Rabeprazol: Estos medicamentos son inhibidores de la bomba de protones (IBP), los cuales se utilizan para disminuir el reflujo gastroesofágico mediante una disminución en la producción de ácido gástrico.
E.-Afectación Muscular y Articular:
- Los corticoides: prednisona, metilprednisolona, pueden ser utilizados en casos de miositis o artritis asociada.
- Metotrexato: en los casos de artritis y fibrosis cutánea.
- AINES: Los antiinflamatorios no esteroideos (AINEs) son útiles para el dolor articular, aunque deben usarse con precaución debido a posibles efectos gastrointestinales.
2.) TRATAMIENTOS BIOLÓGICOS:
A.- Tocilizumab: Anticuerpo monoclonal, anti-receptor de interleucina-6 (IL-6) aprobado por la FDA para el tratamiento de la esclerosis sistémica con enfermedad pulmonar intersticial, mejorando la capacidad ventilatoria del paciente (FVC).
B.-Rituximab: Anticuerpo monoclonal (anti-CD20) que ha mostrado beneficios en la reducción de la fibrosis cutánea y la preservación de la capacidad respiratoria en la esclerosis sistémica con afectación pulmonar.
3.) TRATAMIENTOS AVANZADOS:
- Trasplante de Células Madre Hematopoyéticas (TCMH): Utilizado en pacientes con esclerosis sistémica progresiva que presentan riesgo de fallo orgánico, se ha demostrado que mejora la afección cutánea y la función pulmonar.
4.) NUEVAS TERAPIAS EMERGENTES:
- Los inhibidores de la JAK (Quinasa Janus).
- Anticuerpos monoclonales dirigidos contra TGF-β (factor de crecimiento transformante beta).
Estos dos ultimos estan siendo investigados hoy dia para el tratamiento de la esclerosis sistémica, específicamente para reducir la fibrosis.
En conclusión, como podrán ver, el tratamiento de la esclerosis sistémica (ESc) debe ser multidisciplinario y enfocado en cada caso clínico, y aun no teniendo cura, la meta es mejorar la calidad de vida del paciente.
Saludos,,,
Dr. José Lapenta.
ENGLISH
Scleroderma, also known as systemic sclerosis (SSc) is a disease classified within collagenosis, it is a chronic autoimmune disease characterized by thickening and hardening of the skin and also in some cases of internal organs, caused by an increase in collagen synthesis, which leads to fibrosis of the skin and internal organs (not all cases).
This process is driven by activated fibroblasts and myofibroblasts, by immunological factors that produce an increase in the extracellular matrix, leading to the symptoms described.
The name Scleroderma comes from Greek where "sclero" means hard or stiff, and "derma", skin, so the GREAT DISTINCTIVE CHARACTERISTIC of this disease is the thickening of the skin, which is noted to be tightened by simple touch, sometimes defined as "cardboard skin" or "cardboarding" of the skin, obviously plus other clinical symptoms and laboratory findings that typify this pathology.
ETIOLOGY:
Systemic sclerosis is triggered by genetic and environmental factors.
1.) Environmental factors:
Among the environmental factors that trigger scleroderma, the first is the workers in the SILICA MINES, who, by inhaling the dust, not only develop pulmonary fibrosis, but also develop skin manifestations typical of systemic sclerosis. This fact has been confirmed in various studies (Canada, China and Australia).
2.) Genetic predisposition:
Ethnic groups: in this regard, it has been described or discovered that the African-American population tends to suffer more frequently from this disease in relation to Europeans, also an earlier onset of the disease in which the diffuse type predominates and a greater association with ANTI-topoisomerase I antibodies (SCL-70). Europeans present a greater association with ANTI-centromere antibodies and a milder form of the disease.
HLA antigens: The association of several ALLELES of the Major Histocompatibility System with this disease has been well described in the literature. These are: HLA-DRB111, HLA-DRB111:04, HLA-DPB1, HLA-DPB113:01, HLA-DPB113:01, HLA-DQA1 and HLA-DQB1, HLA-DQA105:01.
We will discuss this topic in our next review entitled HLA AND CONNECTIVE TISSUE DISEASES.
SYMPTOMS:
The symptoms of classic systemic sclerosis (SSc) are divided into:
1.) CUTANEOUS MANIFESTATIONS:
A.- Thickening of the skin: (fibrosis), particularly of the fingers (sclerodactyly) and face, where when the skin hardens, the face takes on an appearance described as "bird face", which is rarely described today. Other cutaneous manifestations include: periungual telangiectasias (typical of systemic sclerosis), calcinosis, poikilodermatous skin, and digital ulcers.
B.- Vascular damage: damage to the endothelial cells of blood vessels, affects fingers and toes, producing the RAYNAUD PHENOMENON (CLICK) which is characterized by changes in skin color due to the effect of cold, which causes vasospasm.
2.) SYSTEMIC MANIFESTATIONS:
Fibrosis or thickening of internal organic tissues mainly causes:
A.- Pulmonary involvement: pulmonary arterial hypertension and interstitial lung disease (interstitial pneumonitis).
B.- Renal involvement: characterized by acute renal failure and hypertension which can be severe.
C.- Cardiac involvement: fibrosis of cardiac tissue can cause myocardial alterations, arrhythmias, and pericardial effusion.
D.- Gastrointestinal involvement: thickening of gastrointestinal tissue can cause: decreased esophageal motility, causing difficulty to swallowing food, gastroesophageal reflux and pseudo intestinal obstruction.
E.- Joint and muscle involvement: arthralgia and myalgia.
F.- General malaise: fatigue.
CLASSIFICATION:
Some dermatological schools speak or classify scleroderma into:
1.) LOCALIZED SCLERODERMA: which is the same:
A.) Localized Scleroderma or Morphea (CLICK), where THERE ARE NO SYSTEMIC SYMPTOMS; in the chapter MORPHEA the terms MORPHEA and LINEAR LOCALIZED SCLERODERMA are well explained.
B.) Localized Scleroderma "En Cup De Sabre": it is a variant of localized scleroderma that manifests ONLY in the frontal region in the form of a depression or linear atrophy that reaches the scalp, giving the appearance of an "axe blow" or "sabre cleft." Some consider it a variant of the so-called progressive hemifacial atrophy. No specific antibodies for scleroderma have been found, nor are there systemic symptoms in most cases.
2.) SYSTEMIC SCLEROSIS (SSc): which is subdivided into three groups today in 2024.
A.- Diffuse cutaneous systemic sclerosis (dcSSc):
It has the worst prognosis, since fibrosis and skin involvement is extensive, practically including almost the entire body, mainly torso and arms. Fibrosis also affects internal organs (heart, kidney, lungs) with rapid progression in early stages of the disease and therefore is the worst of all in terms of survival time.
B.- Limited cutaneous systemic sclerosis (clSSc):
This subtype was known and described as CREST SYNDROME, a variant of limited scleroderma where fibrosis mainly affects the face and arms, and its characteristics are:
C: calcinosis.
R: Raynaud's phenomenon.
E: Sclerodactyly.
S: Esophageal motility disorders.
T: Periungual telangiectasias. (typical)
This variant represents 20% of scleroderma cases, has a better prognosis, its course is slower and is associated with ANTI-centromere antibodies.
Just as DERMATOMYOSITIS (CLICK) without myositis was described, in the scleroderma a variant without cutaneous fibrosis was described and called:
C.-Systemic sclerosis sine scleroderma (ssSSc):
In this variant THERE IS NO CUTANEOUS FIBROSIS, but one or more internal organs are affected and it represents 10% of scleroderma cases. This subtype can be misdiagnosed because it has no skin manifestations, but despite the absence of fibrosis, there may be serious cardiac, renal (sclerodermal crisis) and pulmonary (high blood pressure) manifestations.
For this variant to be diagnosed, in addition to the symptoms described, some of the typical antibodies of scleroderma must be found in the laboratory findings, namely:
Antinuclear antibodies (ANA), Anticentromere antibodies (ACA), Anti-RNA polymerase III antibodies, or Anti-Scl-70 antibodies (topoisomerase I) and others.
DIAGNOSIS:
In 2015, the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) established the following criteria for the diagnosis of scleroderma:
1.) Thickening of the skin of the fingers up to the metacarpophalangeal joints and/or lesions on the tips of the fingers.
2.) Periungual telangiectasias (abnormal capillaries in the nail fold).
3.) Lung involvement.
4.) Raynaud's phenomenon.
5.) Scleroderma-specific autoantibodies:
A.-Antinuclear antibodies (ANA): Present in more than 90% of patients with systemic sclerosis (SSc). Their absence does not exclude the diagnosis. ANA are independent predictors of digital microvascular damage.
B.-Anti-topoisomerase I antibodies (ATA-SCL-70): Associated with diffuse cutaneous systemic sclerosis (dcSSc), fingertip and digital ulcers, and severe lung disease (interstitial pneumonitis).
C.-Anticentromere antibodies (ACA): Associated with limited cutaneous systemic sclerosis (clSSc) and minor pulmonary fibrosis and kidney damage (the former CREST SYNDROME).
D.-Anti-RNA polymerase III antibodies (ARA): Associated with rapid progression of skin thickening, renal crisis, and association with cancer.
E.-Anti-U3 RNP antibodies: Related to pulmonary hypertension, and gastrointestinal complications (Afro-Caribbean patients).
G.-Anti-PM/Scl antibodies: Associated with a high frequency of interstitial lung disease and myopathy.
H.-Anti-Ku antibodies: Associated with OVERLAP polymyositis/systemic sclerosis syndrome, without prominent vascular manifestations.
SUMMARIZING. TODAY 2024, THE CLASSIFICATION OF SYSTEMIC SCLEROSIS IS THE THREE (3) PREVIOUSLY DESCRIBED.
1.) DIFFUSE OR GENERALIZED CUTANEOUS. (dcSSc)
2.) LOCALIZED OR LIMITED CUTANEOUS (clSSc), previous CREST syndrome.
3.) SINE SCLERODERMA (ssSSc), without cutaneous fibrosis.
TREATMENT:
There is NO CURE for systemic sclerosis. Treatments are aimed at improving the specific symptoms of each clinical presentation and are:
1.) MANAGEMENT OF SPECIFIC SYMPTOMS:
A.-Raynaud's phenomenon: nifedipine (calcium antagonists) and phosphodiesterase type 5 (PDE-5) inhibitors, sildenafil and tadalafil to improve circulation and prevent or reduce the frequency of attacks.
-Intravenous iloprost: can be used for severe attacks or digital ulcers.
B.-Lung involvement:
- Cyclophosphamide: Used in severe cases of diffuse scleroderma with lung involvement, although its long-term use is limited by its toxicity.
- Mycophenolate mofetil: like cyclophosphamide, it is used in severe systemic sclerosis with interstitial lung disease.
- Pirfenidone: Although not specifically approved for systemic sclerosis with lung involvement, its use in combination with mycophenolate mofetil is being investigated.
- Nintedanib, an antifibrotic agent, to improve lung function.
- Bosentan, epoprostenol and other prostacyclin analogues: for the treatment of pulmonary hypertension and may also reduce the development of new digital ulcers
C.- Renal Involvement:
- Enalapril, lisinopril: Enzyme inhibitors, Angiotensin-converting angiotensin (ACE) inhibitors are the most recommended for treating scleroderma renal crisis, significantly improving survival.
- Losartan, valsartan: Angiotensin II Receptor Antagonists (ARA-II): These are used as an alternative to ACEIs. They help control blood pressure and can improve management of kidney function.
- Nifedipine and amlodipine: Calcium channel blockers (BCC): useful for improving kidney function, although they are more specific for the treatment of Raynaud's Phenomenon.
D.- Digestive involvement:
- Omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole: These medications are proton pump inhibitors (PPIs), which are used to reduce gastroesophageal reflux by decreasing the production of gastric acid.
E.-Muscle and Joint Involvement:
- Corticosteroids: prednisone, methylprednisolone, can be used in cases of myositis or associated arthritis.
- Methotrexate: in cases of arthritis and skin fibrosis.
- NSAIDs: Nonsteroidal anti-inflammatory drugs (NSAIDs) are useful for joint pain, although they should be used with caution due to possible gastrointestinal effects.
2.) BIOLOGICAL TREATMENTS:
A.- Tocilizumab: Monoclonal antibody, anti-interleukin-6 (IL-6) receptor approved by the FDA for the treatment of systemic sclerosis with interstitial lung disease, improving the patient's ventilatory capacity (FVC).
B.- Rituximab: Monoclonal antibody (anti-CD20) that has shown benefits in reducing skin fibrosis and preserving respiratory capacity in systemic sclerosis with lung involvement.
3.) ADVANCED TREATMENTS:
- Hematopoietic Stem Cell Transplantation (HSCT): Used in patients with progressive systemic sclerosis who are at risk of organ failure, it has been shown to improve skin conditions and lung function.
4.) NEW EMERGING THERAPIES:
- JAK (Janus Kinase) inhibitors.
- Monoclonal antibodies directed against TGF-β (transforming growth factor beta).
These last two are currently being investigated for the treatment of systemic sclerosis, specifically to reduce fibrosis.
In conclusion, as you can see, the treatment of systemic sclerosis (SSc) must be multidisciplinary and focused on each clinical case, and even though there is no cure, the goal is to improve the patient's quality of life.
Greetings...
Dr. José Lapenta R.
EDITORIAL ESPANOL:
====================
Saludos amigos de la red,,, DERMAGIC una vez mas con ustedes,,, LA ESCLERODERMIA, temida enfermedad, asociada como muchas otras enfermedades a los Antígenos de Histocompatibilidad (HLA), de tratamiento difícil y futuro incierto. Estas 59 referencias (1999-2024) muestran algo de las nuevas tendencias diagnósticas y terapéuticas.
PRÓXIMA EDICIÓN: HLA Y COLAGENOSIS .
Saludos,,,
Dr. José Lapenta R.,,,
EDITORIAL ENGLISH:
===================
Greetings friends of the net, DERMAGIC once again with you, THE SCLERODERMA, feared illness, associate as many other illnesses to the Histocompatibility antigens (HLA), of difficult treatment and uncertain future. These 59 references (1999-2024) show something of the new tendencies about diagnose and therapeutic.
NEXT EDITIONS: HLA AND CONNECTIVE TISSUE DISEASES.
Greetings,,,
Dr. José Lapenta R.
=====================================================================
DERMAGIC/EXPRESS(56)
=====================================================================
ESCLEROSIS SISTÉMICA /SYSTEMIC SCLEROSIS
=====================================================================
A.) Current Concepts on the Pathogenesis of Systemic Sclerosis.
B.) Systemic Sclerosis in Adults. Part I: Clinical Features and Pathogenesis.
D.) Occupational and Environmental Scleroderma. Systematic Review and Meta-Analysis.
H.) Association of the HLA-DRB1 With Scleroderma in Chinese Population.
M.) Histological Features of Localized Scleroderma 'En Coup De Sabre': A Study of 16 Cases.
N.) Anti-Pm/SCL-100 and Anti-Rna-Polymerase III Antibodies in Scleroderma.
P.) Systemic Sclerosis-Specific Antibodies: Novel and Classical Biomarkers.
S.) State-of-the-Art Evidence in the Treatment of Systemic Sclerosis.
T.) New Era in Systemic Sclerosis Treatment: Recently Approved Therapeutics.
V.) Systemic Sclerosis in Adults. Part II: Management and Therapeutics.
W.) Advances in Biological and Targeted Therapies for Systemic Sclerosis.
X.) Therapeutic Approaches to Systemic Sclerosis: Recent Approvals and Future Candidate Therapies.
Z.) Emerging Diagnostic and Therapeutic Challenges for Skin Fibrosis in Systemic Sclerosis.
1.)HLA and clinical associations in systemic sclerosis patients with anti-Th/To antibodies.
2.) Heterogenous nuclear RNP C1 and C2 core proteins are targets for an autoantibody found in the serum of a patient with systemic sclerosis and psoriatic arthritis.
3.) Gene expression of types I and III collagen, decorin, matrix metalloproteinases and tissue inhibitors of metalloproteinases in skin fibroblasts from patients with systemic sclerosis.
4.) [Effects of various prokinetic drugs on gastrointestinal transit times in patients with progressive systemic scleroderma]
5.) Fetal-maternal HLA compatibility confers susceptibility to systemic sclerosis.
6.) Skin involvement as a relevant outcome measure in clinical trials of systemic sclerosis.
7.) Epidemiology of systemic sclerosis and related diseases.
AU: Mayes-MD
8.) Type 2 helper T-cell predominance and high CD30 expression in systemic sclerosis.
9.) Impaired quantitative cerebral blood flow in scleroderma patients.
10.) [A patient with systemic scleroderma showing improvement during long-term hemodialysis after renal crisis]
11.) ["Primary" diffuse interstitial fibrosis in coal miners: a new entity? Study Group on Interstitial Pathology of the Society of Thoracic Pathology of the North]
12.) Effect of iloprost infusion on the resistance index of renal vessels of patients with systemic sclerosis.
13.) In situ expression and serum levels of tumor necrosis factor-alpha receptors in patients with early stages of systemic sclerosis.
14.) Anorectal function in systemic sclerosis: correlation with esophageal dysfunction?
15.) Elevated soluble Fas/APO-1 (CD95) levels in silicosis patients without clinical symptoms of autoimmune diseases or malignant tumours.
16.) The coagulation/fibrinolysis balance in systemic sclerosis: evidence for a haematological stress syndrome.
17.) Autoantigen components recognizable by scleroderma sera are exported via ectocytosis of fibroblasts.
18.) Cytokine production in scleroderma patients: effects of therapy with either
iloprost or nifedipine.
18.) Scleroderma, D-penicillamine treatment, and progressive renal failure associated
with positive antimyeloperoxidase antineutrophil cytoplasmic antibodies.
19.) [Spontaneous rupture of the esophagus (Boerhaave syndrome) in a patient with
scleroderma treated by continuous ambulatory peritoneal dialysis]
20.) Association of DM genes in systemic sclerosis is secondary to the association with HLA genes.
21.) Choosing appropriate patients with systemic sclerosis for treatment by autologous
stem cell transplantation.
22.) Development of a protocol for allogeneic marrow transplantation for severe systemic
sclerosis: paradigm for autoimmune disease.
23.) Hypothesis for the pathogenesis of systemic sclerosis.
24.) Hematopoietic stem cell transplantation for autoimmune diseases.
25.) The evolving role of blood and marrow transplantation for the treatment of
autoimmune diseases.
26.) Systemic scleroderma. Multicenter trial of 1 year of treatment with recombinant
interferon gamma [see comments]
26.) [Acupuncture in treatment of inflammatory rheumatic diseases]
27.) Characterization of antinucleolar antibody reactivity in patients with systemic
sclerosis and their relatives.
28.) [Systemic scleroderma and sarcoidosis: 3 new cases]
29.) [Cardiopathy in systemic sclerosis]
30.) Scleroderma and pregnancy.
31.) Epidemiology of systemic sclerosis.
32.) Gastrointestinal features of scleroderma.
33.) Systemic sclerosis in German uranium miners under special consideration of
autoantibody subsets and HLA class II alleles.
34.) DNA allelic alterations within VNTR loci of scleroderma families.
35.) Treatment of systemic sclerosis.
36.) Iloprost effects on phagocytes in patients suffering from ischaemic diseases: in
vivo evidence for down-regulation of alpha M beta 2 integrin.
37.) Association of esophagitis and esophageal strictures with diseases treated with
nonsteroidal anti-inflammatory drugs.
=====================================================================
=====================================================================
1.)HLA and clinical associations in systemic sclerosis patients with anti-Th/To antibodies.
=====================================================================
AU: Falkner-D; Wilson-J; Medsger-TA Jr; Morel-PA
SO: Arthritis-Rheum. 1998 Jan; 41(1): 74-80
AB: OBJECTIVE. To determine the clinical and immunogenetic features of systemic sclerosis (SSc) patients with anti-Th/To autoantibodies. METHODS. HLA class II alleles were determined by DNA oligotyping in a large group of SSc patients with anticentromere antibodies (ACA), anti-topoisomerase I (anti-topo I), and anti-Th/To autoantibodies. RESULTS. Clinical features of the 28 anti-Th/To-positive SSc patients were similar to those observed in the 56 ACA-positive SSc patients, except for a decreased frequency of gastrointestinal involvement in anti-Th/To-positive patients. Immunogenetic analysis revealed a significant increase in the frequency of HLA-DR11 in the anti-Th/To-positive and the anti-topo I-positive patients. The anti-Th/To-positive patients also had a significant reduction in the frequency of HLA-DR7, similar to that seen in ACA-positive SSc patients. CONCLUSION. Despite clinical and immunogenetic similarities with both the ACA- and anti-topo I-positive patients, anti-Th/To-positive SSc patients present a characteristic pattern of clinical and immunogenetic features that may have implications regarding etiology, pathogenesis, and treatment.
=====================================================================
2.) Heterogenous nuclear RNP C1 and C2 core proteins are targets for an autoantibody found in the serum of a patient with systemic sclerosis and psoriatic arthritis.
=====================================================================
AU: Stanek-D; Vencovsky-J; Kafkova-J; Raska-I
SO: Arthritis-Rheum. 1997 Dec; 40(12): 2172-7
AB: OBJECTIVE. To determine a target recognized by anti-Bh autoantibody, found in the serum of a patient with the unusual coexistence of systemic sclerosis (SSc) and psoriatic arthritis (PsA). METHODS. Antigens recognized by the anti-Bh serum were characterized by indirect immunofluorescence on HeLa cells, by conventional immunoblotting using nuclear extract or partially purified preparation of heterogenous nuclear RNP (hnRNP) proteins, and by 2-dimensional immunoblotting. For the analysis of cross-reactivity and immunofluorescence patterns, autoantibodies were affinity-purified by blot elution and then retested. RESULTS. Comparison of the reactivity of the anti-Bh antibody with the monoclonal antibody 4F4 against both the hnRNP C proteins, together with the determination of biochemical properties of the autoantigens, led to the identification of C1 and C2 core proteins as the targets for the anti-Bh autoantibody. CONCLUSION. Several essential components of the spliceosome are targeted by autoantibodies that are present in the sera of patients with systemic rheumatic diseases. We also found that the hnRNP core proteins C1 and C2 are recognized by the autoantibody present in the serum of a patient with SSc and PsA. C1 and C2 hnRNP proteins should be added to the several intracellular autoantigens recently shown to be cleaved by interleukin-1beta-converting enzyme-like enzymes during apoptosis.
=====================================================================
3.) Gene expression of types I and III collagen, decorin, matrix metalloproteinases and tissue inhibitors of metalloproteinases in skin fibroblasts from patients with systemic sclerosis.
=====================================================================
AU: Kuroda-K; Shinkai-H
SO: Arch-Dermatol-Res. 1997 Sep; 289(10): 567-72
AB: Cultured fibroblasts from patients with systemic sclerosis (SSc) and normal individuals were examined for gene expression of types I and III collagen, decorin, matrix metalloproteinases (MMP) MMP-1, MMP-2, and MMP-3, tissue inhibitors of metalloproteinases (TIMP) TIMP-1 and TIMP-2, urokinase- and tissue-type plasminogen activators (u-PA and t-PA). Fibroblasts from patients with early stage SSC (less than 1 year duration of disease) exhibited higher levels of types I and III procollagen, decorin, MMP-1, MMP-3, TIMP-1, and PAs than those from normal individuals. The gene expression of procollagen alpha 1(I) and TIMP-1 mRNAs were increased, but those of decorin, MMP-1, MMP-2, and MMP-3 were decreased, in fibroblasts from SSc patients with mid-stage SSc (2 to 4 years duration) as compared with those from normal individuals. In contrast, no significant difference in gene expression was found between fibroblasts from normal individuals and from patients with late-stage SSc (more than 6 years duration). These results suggest that gene expression of collagen, decorin, and degrading factors is dynamically modulated during fibrillogenesis. The responses of procollagen alpha 1(I) mRNA to IL-1 and TGF-beta were lower in fibroblasts from SSc patients with early and mid-stage disease, but not in those from patients with-late stage disease, than in control fibroblasts, which indicates that these cytokines may be involved in the earlier phases of fibrosis in SSc.
=====================================================================
4.) [Effects of various prokinetic drugs on gastrointestinal transit times in patients with progressive systemic scleroderma]
=====================================================================
AU: Folwaczny-C; Laritz-M; Meurer-M; Endres-SP; Konig-A; Schindlbeck-N
SO: Z-Gastroenterol. 1997 Oct; 35(10): 905-12
AB: The intestine is involved in about half of the cases with progressive-systemic sclerosis. Intestinal transit disturbances which are caused by neuropathy of the enteric nerve system occur frequently. However, upto-date only few studies which determined the effect of prokinetic drugs exist. Patients with intestinal involvement caused by progressive-systemic sclerosis were treated with the prokinetic drugs cisapride (20 mg, TID; n = 9), erythromycin (250 mg, TID; n = 7) and octreotide (50 micrograms s. c., at night time; n = 5) over a period of four weeks. At study entry and after each treatment period the transit times through the stomach, small and large intestine were evaluated by use of the metal-detector test. Gastric emptying was only accelerated by erythromycin (42 +/- 3 min vs. 54 +/- 6 min; p = 0.0422), whereas treatment with cisapride and octreotide did not result in significant changes (48 +/- 4 min; p = 0.3743 and 44 +/- 4 min; p = 0.1975; resp.). Small intestinal transit times were not altered significantly by cisapride (108 +/- 15 min vs. 108 +/- 9 min; p = 0.2733), crythromycin (92 +/- 8 min; p = 0.0707) or octreotide (106 +/- 12 min; p = 0.8927). Furthermore colonic transit was not fastened by none of the prokinetic agents (study entry: 68 +/- 12 h; cisapride: 88 +/- 12 h; p = 0.0569; erythromycin 77 +/- 14 h; p = 0.7349; octreotide 107 +/- 14 h; p = 0.8927). Four patients were withdrawn from the study because of diarrhea. Prokinetic drugs do not seem to have a major impact on intestinal transit times in patients with progressive-systemic sclerosis. The use of these drugs is limited because of frequent side effects.
=====================================================================
5.) Fetal-maternal HLA compatibility confers susceptibility to systemic sclerosis.
=====================================================================
AU: Artlett-CM; Welsh-KI; Black-CM; Jimenez-SA
SO: Immunogenetics. 1998; 47(1): 17-22
AB: Systemic sclerosis (SSc) is a disease of unknown origin, which occurs predominantly in women after childbearing years. There are prominent clinical and histopathologic similarities between SSc and chronic graft-versus-host disease (GVHD). GVHD can occur after blood transfusions or after transplantation with HLA-compatible bone marrow. Here we examined the hypothesis that SSc may be caused by fetal cells crossing the placenta into the maternal circulation and providing donor lymphocytes which recognize disparate HLA antigens, resulting in a reaction similar to chronic GVHD. To test the hypothesis we analyzed the inheritance of HLA class I and class II haplotypes in the families of 37 SSc patients and 42 control individuals. Twenty-six (70.2%) SSc patients had HLA class II alleles compatible either for their offspring or mother, compared with only nine (21%) control individuals. The four patients with juvenile onset SSc we analyzed had alleles compatible with their mothers. These results suggest that in some patients, SSc may, indeed, be a form of chronic GVHD caused by fetal or maternal cells which have crossed the placenta during pregnancy and have remained unrecognized by the host due to class II HLA compatibility, and that subsequent activation of these cells by as yet unknown stimuli result in the development of the disease.
=====================================================================
6.) Skin involvement as a relevant outcome measure in clinical trials of systemic sclerosis.
=====================================================================
AU: Seibold-JR; McCloskey-DA
SO: Curr-Opin-Rheumatol. 1997 Nov; 9(6): 571-5
AB: Advances in our understanding of the pathobiology of scleroderma and the ever-increasing availability of rational candidate therapies have brought the clinical study of scleroderma to the forefront. In the 1990s, consensus measures of outcome have been developed for common disorders such as rheumatoid arthritis. With decisions founded on extensive and validated data, the clinical research community and regulatory agencies have accepted the importance of a demonstration of small degrees of change over relatively short periods of time. The current level of sophistication in scleroderma research is reminiscent of our approach to the study of rheumatoid arthritis in the early 1970s. Accessible, reproducible, and cost-effective measures of outcome are needed in scleroderma. These measures should incorporate clinical meaningfulness and be relevant to quality of life. This review discusses some of the more recent studies of outcome measures in scleroderma, some or all of which are considered relevant to both drug development and clinical care of the individual patients.
=====================================================================
7.) Epidemiology of systemic sclerosis and related diseases.
AU: Mayes-MD
=====================================================================
SO: Curr-Opin-Rheumatol. 1997 Nov; 9(6): 557-61
AB: Epidemiologic studies of scleroderma can provide insight into the dynamics of disease expression over time, over different geographic areas, and over diverse ethnic populations. Although such population studies cannot establish cause and effect relationships, they can reveal associations previously obscured by the relative rarity and clinical diversity of this disease. The incidence rate of systemic sclerosis has been stable over the past 20 years at 19 new cases per million per year. The incidence rate of localized scleroderma or morphea is reported at 27 new cases per million per year and has a benign prognosis overall. Disease expression of systemic scleroderma is influenced by genetic, ethnic, and environmental factors. A cluster of scleroderma cases among Choctaw Native Americans in Oklahoma provides an opportunity to investigate the interaction of genetic and environmental influences. Twin studies suggest a definite but relatively weak genetic component. Case-control studies regarding environmental and occupational exposures have yet to identify risk factors that could serve as triggers for this disease.
=====================================================================
8.) Type 2 helper T-cell predominance and high CD30 expression in systemic sclerosis.
=====================================================================
AU: Mavalia-C; Scaletti-C; Romagnani-P; Carossino-AM; Pignone-A; Emmi-L; Pupilli-C; Pizzolo-G; Maggi-E; Romagnani-S
SO: Am-J-Pathol. 1997 Dec; 151(6): 1751-8
AB: The pattern of cytokine production of skin-infiltrating T cells from patients with progressive systemic sclerosis was investigated. Most CD4+ T-cell clones generated from skin biopsy specimens showed a type 2 helper (Th2) cytokine profile (production of interleukin-4, but no interferon (IFN)-gamma). High interleukin-4 but little or no IFN-gamma mRNA expression was found by in situ hybridization in skin perivascular mononuclear cell infiltrates. The immunohistochemical analysis revealed CD30 expression by high numbers of CD4+ T cells in the same specimens. Finally, the great majority of patients with diffuse disease had elevated levels of soluble CD30 in their sera. These data suggest the existence in patients with progressive systemic sclerosis of a predominant activation of Th2-like T cells, which may account for the major alterations (endothelial cell injury, fibrosis, and autoantibody production) occurring in this disease.
=====================================================================
9.) Impaired quantitative cerebral blood flow in scleroderma patients.
=====================================================================
AU: Nobili-F; Cutolo-M; Sulli-A; Castaldi-A; Sardanelli-F; Accardo-S; Rosadini-G; Rodriguez-G
SO: J-Neurol-Sci. 1997 Nov 6; 152(1): 63-71
AB: Systemic Sclerosis (SSc) is a multisystem disease characterised by proliferation of vascular tissue, obliterative microvascular lesions and diffuse organ fibrosis. Despite widespread vascular disease, Central Nervous System complaints are only infrequently reported and it is uncertain whether they merely derive from systemic complications or whether they may be also caused by a primary vascular process within the brain. Regional cerebral blood flow (rCBF) was quantitatively measured by the 133Xenon clearance technique in twenty-seven consecutive SSc patients without relevant systemic complications and with different severity of vascular involvement, as staged by nailfold capillary videomicroscopy (NCV). Absolute, percent, and asymmetry rCBF values were compared (z-statistics) with age- and sex-matched healthy controls. Cerebral MRI and Mini-Mental State Examination (MMSE) were also performed. Doppler sonography of neck vessels and Transcranial Doppler sonography (TCD) were performed in patients presenting rCBF reduction. Cerebral hypoperfusion was found in the 52% of patients, i.e.: in 33% of patients with the 'early' NCV pattern, in 56% of patients with the 'active' pattern, and in 67% of patients with the 'late' NCV pattern. Thirty percent were the MRIs showing focal and/or diffuse signal abnormalities in the white matter of both hemispheres with the highest rate (44%) in the 'late' NCV pattern. MMSE disclosed mild dementia in one patient in the 'late' NCV group and some mistakes in 6 more patients, in the 'active' or 'late' NCV groups, whereas TCD failed to find significant stenosis of Willis' arteries. Cerebral hypoperfusion is shown for the first time in a substantial part of SSc patients without either neurological symptoms or relevant systemic complications. It is suggested that the rCBF reduction might be related to the systemic scleroderma microangiopathy although, probably due to the paucity of connective tissue in cerebral vessels, the vast majority of patients remains in a subclinical phase.
=====================================================================
10.) [A patient with systemic scleroderma showing improvement during long-term hemodialysis after renal crisis]
=====================================================================
AU: Fuse-Y; Muramatsu-M; Sugiyama-S; Maeda-K
SO: Ryumachi. 1997 Aug; 37(4): 574-80
AB: A 68-year-old man experienced systemic pruritus since he was 63 years old, and systemic sclerosis and skin pigmentation were observed when he was 64. When he developed dyspnea the same year, he was admitted and SSc was diagnosed on the basis of the clinical and skin biopsy findings, lung fibrosis on X-P and TBLB findings. At 65, his dyspnea reappeared along with elevated blood pressure, acute renal failure and lung congestion, and he was diagnosed as having a scleroderma renal crisis (SRC) from the clinical and renal biopsy findings. Hemodialysis was started because he showed mental disturbance, and this and other acute symptoms were subsequently reduced. As he showed no recovery from his renal failure, the patient has been maintained on hemodialysis for over four years now. In the meantime, his sclerosis has improved and antinuclear antibody almost disappeared. Hemodialysis appears to be the most likely reason for his improvement, although spontaneous remission, D-penicillamine and angiotensin converting enzyme (ACE) inhibitor therapy may also have contributed, considering the short period and the small amount of drugs given until improvement.
=====================================================================
11.) ["Primary" diffuse interstitial fibrosis in coal miners: a new entity? Study Group on Interstitial Pathology of the Society of Thoracic Pathology of the North]
=====================================================================
AU: Brichet-A; Wallaert-B; Gosselin-B; Remy-Jardin-M; Voisin-C; Lafitte-JJ; Tonnel-AB
SO: Rev-Mal-Respir. 1997 Sep; 14(4): 277-85
AB: It is well known that silica exposure leads in an experimental model to the development of an acute fibrotic process. In human beings two main observations have already been done: (1) silica exposure is frequently associated with the development of connective tissue disease (CTD), especially progressive systemic sclerosis; (2) 10 to 20% patients with CTD developed pulmonary fibrosis. In this context we report 26 cases of coal miners who presented with clinical, radiological, biological and functional characteristics mimicking idiopathic pulmonary fibrosis (IPF), with or without associated coal worker's pneumoconiosis (CWP). All were men; mean age was 68 +/- 9.2 years. Twenty-three were smokers. Duration of exposure was 28.8 +/- 9.1 years. All the patients had dyspnea (stage III, IV in the NHYA classification) and diffuse crackles. Eleven out of 26 had finger clubbing. Computed tomography showed honeycombing (23 cases), and/or ground glass opacities (6 cases) with bronchiectasis (3 cases) predominant in the lower lobes; 19 had radiological signs of CWP, micronodules (n = 16) and nodules (n = 3) predominant in the upper lobes. BAL exhibited an increased % of neutrophils (11.9 +/- 16.1%). Lung function demonstrated a restrictive pattern (TLC = 73 +/- 15.6% and VC = 80 +/- 18% of predicted values) associated with a decreased DLCO (51.8 +/- 23.6% of predicted values) and hypoxemia (at rest = 66.5 +/- 11.2 mmHg, upon effort = 56 +/- 12 mmHg). Lung biopsies were performed in four cases and demonstrated interstitial fibrosis of intraalveolar septum with an accumulation of immune and inflammatory cells similar to the one described in IPF. The association between IPF and silica exposure with or without associated CWP points out the problem of legal recognition of idiopathic-like pulmonary fibrosis as a complication of the occupational exposure of coal workers.
=====================================================================
12.) Effect of iloprost infusion on the resistance index of renal vessels of patients with systemic sclerosis.
=====================================================================
AU: Scorza-R; Rivolta-R; Mascagni-B; Berruti-V; Bazzi-S; Castagnone-D; Quarto-di-Palo-F
SO: J-Rheumatol. 1997 Oct; 24(10): 1944-8
AB: OBJECTIVE: To investigate the effect of iloprost, a stable prostacycline analog, on kidney blood flow in patients with systemic sclerosis (SSc), using color flow Doppler sonography. METHODS: The acute effect of the drug was studied in 10 patients with SSc with elevated resistance index (RI) levels (all RI values reported are multiplied by 100). Iloprost was administered intravenously (2 ng/kg/min for a period of 8 h). To study the effects of chronic drug administration, 16 patients with SSc were randomly assigned to 2 groups of 8 cases each. The first group was treated with 9 infusions of iloprost in 6 mo. The second group was treated with slow release nifedipine (40 mg/day) for 6 mo. RESULTS: Interlobar artery RI (median 67 vs 61; p = 0.02) and cortical vessel RI (median 65 vs 54; p = 0.001) were reduced after acute treatment. In chronic drug administration, RI values were not modified by nifedipine, while iloprost reduced the RI of the interlobar (median 69 vs 61; p < 0.03) and cortical arteries (median 66 vs 58: p < 0.01). CONCLUSION: Our findings suggest iloprost might be useful for treatment of scleroderma renal vasospasm.
=====================================================================
13.) In situ expression and serum levels of tumor necrosis factor-alpha receptors in patients with early stages of systemic sclerosis.
=====================================================================
AU: Gruschwitz-MS; Albrecht-M; Vieth-G; Haustein-UF
SO: J-Rheumatol. 1997 Oct; 24(10): 1936-43
AB: OBJECTIVE: Tumor necrosis factor-alpha (TNF-alpha) is an important cytokine in the early stage of systemic sclerosis (SSc), which is characterized by mononuclear cell infiltration and microvascular alterations. Most effects of TNF-alpha are mediated by its interaction with 2 types of TNF receptors and depend on their surface expression on individual cell subsets. Our purpose was to correlate the serum levels of soluble TNF receptors-TNF-RI(p55) and RII(p75)-with (1) their in situ expression and distribution in lesional skin and on peripheral blood mononuclear cells (PBMC), and (2) the clinical disease progression and inflammatory serum variables in patients with SSc. METHODS: Serum samples of 32 patients with SSc and 36 healthy probands were examined by ELISA. We performed immunohistological stainings and in situ hybridization on cryostat sections of skin lesions, cytometric analysis on PBMC, and reverse transcriptase polymerase chain reactions using RNA from cultured skin fibroblasts in 17 of these 36 patients. RESULTS: In contrast to healthy skin and chronic fibrotic SSc, TNF-RI is expressed on about 30% of mononuclear infiltrating cells in early skin lesions. Neither TNF-RI nor RII was detectable on fibroblasts by immunohistochemistry, but specific mRNA could be found on the transcriptional level. TNF-RII is found on most lymphocytes and on 30-50% of endothelial cells, especially in early SSc. Expression of both receptor types on PBMC in patients and controls was not significantly different. Serum levels of soluble TNF-RI and RII correlated well with their in situ expression and with clinical and laboratory signs of inflammation and disease progression in patients with SSc. CONCLUSION: Our data provide evidence for a central role of the TNF-alpha/TNF-R system in the early pathological events of scleroderma with prominent inflammation and endothelial cell damage. Determination of TNF-R serum levels provides a useful diagnostic tool for characterization of the disease stage and progression, and to guide experimental therapy in patients with SSc.
=====================================================================
14.) Anorectal function in systemic sclerosis: correlation with esophageal dysfunction?
=====================================================================
AU: Lock-G; Zeuner-M; Lang-B; Hein-R; Scholmerich-J; Holstege-A
SO: Dis-Colon-Rectum. 1997 Nov; 40(11): 1328-35
AB: PURPOSE: This study was designed to compare esophageal and anorectal function parameters in patients with systemic sclerosis and to define the role of anorectal manometry in the diagnosis of gastrointestinal involvement of systemic sclerosis. PATIENTS AND METHODS: Twenty-six consecutive patients (22 females) with systemic sclerosis originally referred for assessment of esophageal function were evaluated by esophageal and anorectal manometry. Anorectal function parameters were compared between patients with normal and those with disturbed esophageal function. RESULTS: A total of 17 of 26 patients (65 percent) had severe esophageal dysfunction with aperistalsis of the lower two-thirds of the esophagus, whereas 9 patients (35 percent) had normal esophageal manometry. Only three patients (11.5 percent) suffered from occasional fecal incontinence. Anorectal function parameters (resting pressure, maximum squeeze pressure, perception threshold) were not significantly different between patients with normal and those with disturbed esophageal motility. Rectoanal inhibitory reflex was excitable in nearly 90 percent of patients. CONCLUSION: In an unselected group of patients with systemic sclerosis, fecal incontinence and abnormal anorectal function are rather rare findings. Anorectal manometry cannot differentiate between patients with and without gastrointestinal involvement of systemic sclerosis.
=====================================================================
15.) Elevated soluble Fas/APO-1 (CD95) levels in silicosis patients without clinical symptoms of autoimmune diseases or malignant tumours.
=====================================================================
AU: Tomokuni-A; Aikoh-T; Matsuki-T; Isozaki-Y; Otsuki-T; Kita-S; Ueki-H; Kusaka-M; Kishimoto-T; Ueki-A
SO: Clin-Exp-Immunol. 1997 Nov; 110(2): 303-9
AB: Soluble Fas (sFas) is produced as translation products of alternative mRNA splicing, and antagonizes the membranous Fas molecule in Fas/Fas ligand interactions. We investigated the serum sFas levels in 64 Japanese silicosis patients with no clinical symptoms of autoimmune diseases or malignant tumours, using ELISA for sFas. The serum sFas levels in the silicosis patients were significantly higher than those in healthy volunteers. Elevated serum sFas levels were also detected in patients with systemic lupus erythematosus but, unexpectedly, no difference was observed in sFas levels between progressive systemic sclerosis patients and healthy volunteers. On the other hand, there was no significant difference in the expression of Fas on peripheral blood lymphocytes between the patients with silicosis and age-matched healthy volunteers. These observations provided the first evidence that serum sFas levels are elevated in silicosis patients without clinical symptoms of autoimmune diseases or malignant tumours. It remains to be clarified whether patients with elevated sFas levels have a tendency to develop autoimmune diseases later, or whether some other distinct factor(s) is necessary to initiate the progression of autoimmune diseases.
=====================================================================
16.) The coagulation/fibrinolysis balance in systemic sclerosis: evidence for a haematological stress syndrome.
=====================================================================
AU: Ames-PR; Lupoli-S; Alves-J; Atsumi-T; Edwards-C; Iannaccone-L; Khamashta-MA; Hughes-GR; Brancaccio-V
SO: Br-J-Rheumatol. 1997 Oct; 36(10): 1045-50
AB: Systemic sclerosis (SSc) is a disease characterized by progressive microvascular occlusion and fibrosis resulting in irreversible organ damage, the pathogenesis of which is felt to be of vascular origin. To gain a comprehensive view of the coagulation/fibrinolytic balance in SSc, a number of haemostatic and fibrinolytic variables were measured in 26 SSc patients (11 limited, 15 diffuse) and in 22 control subjects. Of the coagulation activation markers, the mean plasma level of prothrombin fragment 1 + 2 (F1 + 2), but not of thrombin-antithrombin complexes (TAT), was higher in SSc patients than in controls (P < 0.001). Plasma levels of fibrin split product D-dimer (DD), fibrinogen (FNG) and von Willebrand factor (vWF) were higher amongst patients than controls (P < 0.001). vWF and FNG levels were positively correlated (P < 0.001). Mean levels of DD and vWF were more elevated in patients with diffuse than limited disease (P = 0.001 and P = 0.04, respectively). On the fibrinolytic side, defective tissue plasminogen activator (tPA) release (venous occlusion test, stimulated level < basal level) was noted in 46% (12/26) of SSc patients, but only in 4% (1/22) of controls. Patients had higher mean levels of tPA inhibitor (PAI) than controls (P < 0.001), levels being more elevated amongst patients with diffuse than limited disease (P = 0.01). An abnormally high lipoprotein (a) [Lp(a)] level was found in 9% (2/20) of control subjects, but in 30% (8/26) of SSc patients (P = 0.04) where it clustered with fibrinolytic defects. Altogether, these data suggest that patients with SSc are in a hypercoagulable state characterized by elevated plasma levels of FNG and vWF, by a dual hypofibrinolytic pattern (defective tPA release and elevated PAI), and by increased thrombin generation with enhanced fibrin formation. Higher levels of vWF, DD and PAI in patients with diffuse disease are consistent with more extensive (micro)vascular involvement, although no causal relationship can be inferred. The lack of a parallel increase of TAT with F1 + 2, in the presence of normal levels of antithrombin III (ATIII), indirectly suggests an impairment of the heparan sulphate-ATIII system which would favour thrombin generation. Since thrombin may act as a mitogen for fibroblasts, may upregulate vWF, PAI and endothelin production by endothelial cells, and may promote fibrin deposition on the vessel wall leading to worsening of microvascular occlusions, limitation of thrombin generation, besides fibrinolytic enhancement, could represent a possible coadjuvant interventional strategy.
=====================================================================
17.) Autoantigen components recognizable by scleroderma sera are exported via ectocytosis of fibroblasts.
=====================================================================
AU: Hsu-TC; Lee-TL; Tsay-GJ
SO: Br-J-Rheumatol. 1997 Oct; 36(10): 1038-44
AB: Previously, we have demonstrated that ectocytosis, a unique cell trafficking process to export a specific subset of cellular proteins in the form of membrane vesicles, can be triggered from human skin fibroblasts cultured in a three-dimensional collagen lattice upon stress relaxation. The same culturing system was employed in the present study using fibroblasts isolated from patients with systemic sclerosis (SSc). To see whether any putative intracellular autoantigens causing SSc might escape out of cells by way of ectocytosis, the same stress-relaxation method was used to induce a synchronized ectocytosis among cultured cells. Membrane vesicles released by scleroderma fibroblasts were subsequently isolated, resolved on SDS-PAGE and immunoblotted with sera from 89 patients with various autoimmune diseases and 11 normal volunteers. Three major polypeptides with apparent mol. wts of 12-14, 32-34 and 70-80 kDa are prominent bands on both SDS-PAGE and immunoblots. The 32-34 kDa polypeptide has been further identified as a member of the annexin protein family, while the 70-80 kDa protein has been shown to be topoisomerase I, as judged by its reactivity to patients' sera and a rabbit polyclonal antibody, and as also judged by a functional assay. In conclusion, our results suggest that ectocytosis might be one of the potential pathways for cells to export intracellular antigens and subsequently cause autoimmune reactions.
=====================================================================
18.) Cytokine production in scleroderma patients: effects of therapy with either
iloprost or nifedipine.
=====================================================================
Della Bella S; Molteni M; Mascagni B; Zulian C; Compasso S; Scorza R
Institute of Internal Medicine, Infectious Diseases and Immunopathology, IRCCS
Ospedale Maggiore di Milano, Italy.
Clin Exp Rheumatol (ITALY) Mar-Apr 1997 15 (2) p135-41 ISSN: 0392-856X
Language: ENGLISH
Document Type: CLINICAL TRIAL; JOURNAL ARTICLE; RANDOMIZED CONTROLLED TRIAL
Journal Announcement: 9711
Subfile: INDEX MEDICUS
OBJECTIVE: To compare the long-term effects of intermittent infusion of iloprost
with those of oral nifedipine on the in vitro production of cytokines in patients
with systemic sclerosis (SSc), and to evaluate their relationship with the effects of
the two treatments on clinical parameters. METHODS: The production of cytokines by
alloactivated circulating mononucleated cells was assessed before and after one year
of treatment in a subset of 31 patients enrolled in a 12-month randomized clinical
trial. Nineteen patients were treated with a 5-day (8 hr per day), 2.0 ng/kg per
minute infusion followed by a 1-day infusion every 6 weeks; 12 patients were treated
with an oral slow-release formulation of nifedipine, 20 mg twice daily. Quantitative
determinations of interleukin-1 beta (IL1-beta) and interleukin-6 (IL6) in the
culture supernatants were performed with a commercial ELISA; the levels of tumor
necrosis factor-alpha (TNF-alpha), and interferon-gamma (IFN-gamma) were measured by
specific radioimmunometric assays. RESULTS: The production of IL1-beta was
significantly lower in the iloprost group than in the nifedipine group. Both the
cutaneous fibrosis and the capillaroscopic patterns were better in patients treated
with iloprost than in patients treated with nifedipine. There was a significant
positive covariance between IL1-beta changes and the changes in both the skin score
and the capillaroscopic score. CONCLUSION: There are several mechanisms by which
iloprost could exert its clinical efficacy. Vasodilatation and inhibition of
platelet aggregation are certainly important, but they are transient. We suggest
that the long-lasting modulation of the cytokine network observed in the present
study could be another potential mechanism responsible for the persistent efficacy of
iloprost despite its intermittent administration.
=====================================================================
18.) Scleroderma, D-penicillamine treatment, and progressive renal failure associated
with positive antimyeloperoxidase antineutrophil cytoplasmic antibodies.
=====================================================================
Hillis GS; Khan IH; Simpson JG; Rees AJ
Department of Medicine, University of Aberdeen, Scotland.
Am J Kidney Dis (UNITED STATES) Aug 1997 30 (2) p279-81 ISSN: 0272-6386
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9711
Subfile: INDEX MEDICUS
Progressive renal failure in patients with scleroderma is a sinister development
that is usually attributed to impaired renal blood flow. In some exceptional cases,
the underlying pathology is a crescentic glomerulonephritis, which has been
associated with positive antineutrophil cytoplasmic antibodies, and in particular
antimyeloperoxidase antibodies. The prognosis in such cases has been very poor. We
report such a patient whose renal function has improved and stabilized on
immunosuppressive therapy.
=====================================================================
19.) [Spontaneous rupture of the esophagus (Boerhaave syndrome) in a patient with
scleroderma treated by continuous ambulatory peritoneal dialysis]
=====================================================================
Rupture spontanee de l'oesophage (syndrome de Boerhaave) chez une patiente
sclerodermique traitee par dialyse peritoneale continue ambulatoire.
Level C; de Precigout V; Lasseur C; Hachem D; Berge F; Larroumet N; Carles J;
Blanchetier V; Videau J; Combe C; Aparicio M
Service de nephrologie, hopital Pellegrin, Bordeaux, France.
Rev Med Interne (FRANCE) Jul 1997 18 (7) p566-70 ISSN: 0248-8663
Language: FRENCH Summary Language: ENGLISH
Document Type:
JOURNAL ARTICLE; REVIEW; REVIEW OF REPORTED CASES English Abstract
Journal Announcement: 9710
Subfile: INDEX MEDICUS
Esophageal involvement is a common situation found in 50 to 80% of patients with
scleroderma, but Boerhaave's syndrome is rare in this context. The authors report
the first case of spontaneous esophageal rupture occurring in a chronic renal failure
patient treated by continuous ambulatory peritoneal dialysis. In this observation,
sclerodermal esophageal dyskinesia, chronic renal failure which is a classical cause
of vomiting and the peritoneal dialysis which play an increasing role in the
intraabdominal pressure are potential contributing factors to Boerhave's syndrome.
In such patients presenting risk factors, even if they are asymptomatic, it seems
reasonable to propose esophageal explorations with manometry or/and endoscopy looking
for dyskinesia or other complications of gastro-esophageal reflux. (20 References)
=====================================================================
20.) Association of DM genes in systemic sclerosis is secondary to the association with HLA genes.
=====================================================================
Takeuchi F; Takizawa K; Nabeta H; Kuwata S; Ito K
Department of Internal Medicine and Physical Therapy, Faculty of Medicine,
University of Tokyo, Japan.
Scand J Rheumatol (NORWAY) 1997 26 (3) p174-9 ISSN: 0300-9742
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9710
Subfile: INDEX MEDICUS
The contribution of polymorphism of DMA and DMB alleles to the pathogenesis of
Japanese Systemic Sclerosis (SSc) was studied in 55 Japanese SSc patients and 77
normal Japanese subjects using the PCR-RFLP (restriction fragment length
polymorphism) method. The allele frequencies of DMB*0101 allele were increased in
SSc with diffuse scleroderma (70.0% vs 49.4%, p < 0.05, pc = not significant (NS))
and in SSc with antitopoisomerase I antibody (a-Scl-70), (68.2%, p < 0.05, pc = NS).
The phenotype frequencies of DMB*0101 in these subgroups of SSc were increased
significantly (95.0%, p = 0.014, pc < 0.05; 95.5%, p = 0.0088, pc < 0.05,
respectively). In contrast, DMB*0102 and DMB*0103 alleles tended to decrease in
diffuse scleroderma and SSc with a-Scl-70, but the decreases were not significant.
Association analysis among DMA, DMB, and DRB1*1502 in Japanese SSc with diffuse
scleroderma and SSc with a-Scl-70 indicated that the increase in DMA*0101 was not
primary, but reflected an increase in HLA DRB1*1502.
=====================================================================
21.) Choosing appropriate patients with systemic sclerosis for treatment by autologous
stem cell transplantation.
=====================================================================
Clements PJ; Furst DE
University of California at Los Angeles School of Medicine, Department of Medicine,
USA.
J Rheumatol Suppl (CANADA) May 1997 48 p85-8 ISSN: 0380-0903
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9709
Subfile: INDEX MEDICUS
Systemic sclerosis (SSc) with diffuse cutaneous scleroderma and visceral organ
involvement is associated with considerable morbidity and mortality almost from its
inception. Since the risk of accrual and progression of skin and organ complications
is greatest in the first few years of SSc, the best opportunity for significantly
modifying the course of SSc (prolonging survival and/or preventing or lessening the
progressivity of organ involvement) is probably limited to the first 3 to 4 years.
Transplantation of autologous stem cells (ASCT), after immune ablation of the host,
has the potential to modify the disease course. Even though the mortality risk of
ASCT is low (< 2% mortality in the first year), the mortality risk of the disease
being treated must justify that risk. We suggest that patients with diffuse Sc of
short duration (< 3 years from the first non-Raynaud sign/symptom) with evidence of
at least mild involvement of heart, lung, or kidney, have sufficiently severe disease
to warrant ASCT. In contrast, we suggest that patients with severe/end stage organ
involvement have progressed to the point where ASCT will not be helpful in improving
that degree of organ involvement. (13 References)
=====================================================================
22.) Development of a protocol for allogeneic marrow transplantation for severe systemic
sclerosis: paradigm for autoimmune disease.
=====================================================================
Nash RA; McSweeney PA; Storb R; Nelson JL; Gauthier J; Furst DE; Sullivan KM
Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA
98104, USA.
J Rheumatol Suppl (CANADA) May 1997 48 p72-8 ISSN: 0380-0903
Contract/Grant No.: CA18029--CA--NCI; CA15704--CA--NCI; CA18221--CA--NCI; +
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9709
Subfile: INDEX MEDICUS
Some types of severe autoimmune disease are associated with significant morbidity
and a high mortality rate. Many of these cases occur in young adults who, even if
they survive, become severely debilitated. Systemic sclerosis (SSc) is a paradigm
for other severe autoimmune diseases in which patients with poor prognostic features
can be identified early in the course of the disease. Allogeneic marrow
transplantation may be effective for the control of autoimmune diseases like SSc
because the preparative regimen will significantly suppress the host immune system
and the antihost effects of the donor immune system in the engrafted marrow will help
maintain the suppression of the host immune system. Considering the morbidity and
poor prognosis associated with severe SSc and the favorable outcome now associated
with allogeneic marrow transplantation from HLA identical siblings for other
nonmalignant diseases, Phase I and II studies are warranted. These will evaluate the
safety of allogeneic marrow transplantation and explore its role in the management
and control of a severe autoimmune disease. We review issues important in the
development of an allogeneic marrow transplant protocol for severe SSc, including
patient selection, plan of treatment, prevention of graft versus host disease,
supportive care, and evaluation after transplant. (51 References)
=====================================================================
23.) Hypothesis for the pathogenesis of systemic sclerosis.
=====================================================================
Furst DE; Clements PJ
Section of Rheumatology and Immunology, Virginia Mason Research Center, Seattle,
Washington 98101, USA.
J Rheumatol Suppl (CANADA) May 1997 48 p53-7 ISSN: 0380-0903
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9709
Subfile: INDEX MEDICUS
An hypothesis for the pathogenesis of systemic sclerosis is introduced. It posits
a genetic background with environmental stimuli that activate immune cells. The
immune cells, in turn, may damage vascular endothelium, cause proliferation of
fibroblasts, or stimulate fibroblasts to produce collagen. Endothelial cell damage
can also activate the immune system or induce fibroblast proliferation. Associated
with fibroblast proliferation may be immune activation or collagen production.
Finally, collagen production and end organ damage can induce immune activation thus
perpetuating the cycle. Raynaud's phenomenon, an early finding in systemic sclerosis
can cause vascular damage, thus entering the cycle at a different point than other
environmental stimuli. This hypothesis will undoubtedly require change as data
emerge, but it presents a conceptual model for testing and modification. (74
References)
=====================================================================
24.) Hematopoietic stem cell transplantation for autoimmune diseases.
=====================================================================
Brooks PM
Department of Medicine, University of New South Wales, St. Vincent's Hospital,
Darlinghurst, Sydney, Australia.
J Rheumatol Suppl (CANADA) May 1997 48 p19-22 ISSN: 0380-0903
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9709
Subfile: INDEX MEDICUS
Autoimmune diseases such as systemic lupus erythematosus, scleroderma, and
rheumatoid arthritis cause significant morbidity and mortality. Although aggressive
treatments may suppress disease activity in some cases, there are few if any complete
cures. Since these conditions arise as a direct result of dysregulation of the
immune system, modification of immune stem cells may be important in their control.
Some slow acting antirheumatic drugs have significant effect on bone marrow, and more
recently a number of case reports have appeared in which autoimmune diseases have
gone into remission after bone marrow transplantation for other reasons. Data from
animal models of autoimmune disease show significant abrogation of inflammation
following bone marrow transplantation. Advances in the technology of stem cell
transplantation coupled with increasing ability to identify at an early stage those
patients likely to develop severe autoimmune disease require an indepth study of the
role of stem cell transplantation for these conditions. (22 References)
=====================================================================
25.) The evolving role of blood and marrow transplantation for the treatment of
autoimmune diseases.
=====================================================================
Sullivan KM; Furst DE
Program in Long-Term Follow-Up, Fred Hutchinson Cancer Research Center, USA.
J Rheumatol Suppl (CANADA) May 1997 48 p1-4 ISSN: 0380-0903
Contract/Grant No.: HL36444--HL--NHLBI; CA18221--CA--NCI; CA18029--CA--NCI; +
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9709
Subfile: INDEX MEDICUS
With over 4 decades of seminal contributions to the development and application of
BMT, Dr. Thomas stresses the importance of collaboration between rheumatologists and
transplant clinicians in developing this evolving area of treatment. While the
debate concerning the value of TBI in the conditioning regimen and the use of
autologous or allogeneic stem cells will continue, he states there is simply no other
way to answer these questions than to begin well designed clinical studies. As
pointed out by Dr. Hahn, unexpected post-transplant complications may arise in
patients with SSc and SLE and possibly require modifications to the transplant
procedure similar to the experience in patients with other specific diseases. Other
difficulties may be encountered, including restricted funding of the transplant
procedure by insurance carriers. The emergence of managed care contracts and payer
limitations in the United States described by Dr. Appelbaum could hinder the
development of innovative, curative therapies. As initial clinical data are being
collected, it is vital to actively support patient referral and participation in
clinical studies that will ultimately establish the indications, risks, costs, and
benefits of hematopoietic stem cell transplantation for autoimmune disease. (59
References)
=====================================================================
26.) Systemic scleroderma. Multicenter trial of 1 year of treatment with recombinant
interferon gamma [see comments]
=====================================================================
Hunzelmann N; Anders S; Fierlbeck G; Hein R; Herrmann K; Albrecht M; Bell S; Thur J;
Muche R; Adelmann-Grill B; Wehner-Caroli J; Gaus W; Krieg T
Department of Dermatology, University of Cologne, Germany.
Arch Dermatol (UNITED STATES) May 1997 133 (5) p609-13 ISSN: 0003-987X
Note: Comment in: Arch Dermatol 1997 May;133(5 ):637-42
Language: ENGLISH
Document Type: CLINICAL TRIAL; JOURNAL ARTICLE; MULTICENTER STUDY
Journal Announcement: 9708
Subfile: AIM; INDEX MEDICUS
OBJECTIVE: To confirm significant improvement of the skin score in systemic
sclerosis by treatment with interferon gamma in a larger group of patients and to
investigate on a molecular level the influence of interferon gamma on collagen type I
messenger RNA expression. DESIGN: Open, noncontrolled multicenter study. SETTING:
Five outpatient clinics specializing in the care of systemic scleroderma. PATIENTS:
Thirty-two patients suffering from the diffuse or limited form of systemic sclerosis
and progressive disease were recruited; 20 patients finished the study.
INTERVENTION: Each patient received interferon gamma, 50 micrograms subcutaneously 3
times a week for 1 year. MAIN OUTCOME MEASURE: Skin score, collagen type I messenger
RNA in skin biopsy specimens. RESULTS: The patients who completed the study showed
an unchanged median skin score after 1 year of therapy. In addition, similar
collagen type I messenger RNA levels were detected in skin biopsy specimens taken
from involved skin before and after therapy in these patients. CONCLUSIONS:
Treatment of systemic scleroderma with interferon gamma is associated with
stabilization of the skin score and lack of worsening of visceral involvement.
=====================================================================
26.) [Acupuncture in treatment of inflammatory rheumatic diseases]
Akupunktur bei der Behandlung entzundlich-rheumatischer Erkrankungen.
=====================================================================
Lautenschlager J
Medizinische Hochschule Hannover Abteilung Rheumatologie.
Z Rheumatol (GERMANY) Jan-Feb 1997 56 (1) p8-20 ISSN: 0340-1855
Language: GERMAN Summary Language: ENGLISH
Document Type:
JOURNAL ARTICLE; META-ANALYSIS English Abstract
Journal Announcement: 9708
Subfile: INDEX MEDICUS
Seventeen studies were examined with regard to efficacy and scientific quality of
acupuncture in rheumatoid arthritis, spondarthropathy, lupus erythematosus, local and
progressive systemic scleroderma. Acupuncture cannot be recommended for treatment of
these diseases. By far, the most studies examined failed to show sufficient quality.
Tags: Human
Descriptors: *Acupuncture Therapy; *Arthritis, Rheumatoid--Therapy--TH; *Lupus
Erythematosus, Systemic--Therapy--TH; *Scleroderma, Circumscribed--Therapy--TH;
*Scleroderma, Systemic--Therapy--TH; *Spondylitis, Ankylosing--Therapy--TH; Quality
Assurance, Health Care; Treatment Outcome
=====================================================================
27.) Characterization of antinucleolar antibody reactivity in patients with systemic
sclerosis and their relatives.
=====================================================================
Harvey G; Black C; Maddison P; McHugh N
Bath Institute for Rheumatic Diseases, England.
J Rheumatol (CANADA) Mar 1997 24 (3) p477-84 ISSN: 0315-162X
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9708
Subfile: INDEX MEDICUS
OBJECTIVE: To study the prevalence and specificity of antinucleolar antibodies
(ANoA) in patients with systemic sclerosis (SSc), their spouses, and their first-
degree relatives, and to investigate whether SSc family members have greater
frequency of ANoA than expected. METHODS: The sera of 58 SSc probands, 4 first-
degree relatives with SSc, 215 first-degree relatives without SSc, and 24 spouses
were screened for ANoA by indirect immunofluorescence (IF), and nucleolar antigens
were characterized by immunoprecipitation (IP) of 35S methionine labeled K562 cell
extracts. Sera from 118 randomly chosen family members without SSc were separately
compared with 120 age and sex matched blood donor controls. RESULTS: Antinucleolar
reactivity was detected by IF in 25 patients with SSc (40.3%), in 33 non-SSc
relatives (15.3%), and in 4 spouses (16.7%). Twenty-four sera had autoantibodies to
defined nucleolar antigens by IP (seven Pm-Scl, ten RNA polymerase (pol) I, four U3
RNP, three Th RNP), and all were from patients with SSc (38.7%). No serum had more
than one type of nucleolar-specific autoantibody. Four sera had autoantibodies to
topoisomerase I (topo I) and RNA pol II, one of which also recognized RNA pol I and
RNA pol III. Antinucleolar IF was significantly more common in the unaffected first-
degree relatives of patients with SSc (18.1%) than in controls (8.3%; p < 0.05). A
small number of sera from both relatives and controls recognized bands by IP, none of
which was identified as a SSc-specific autoantigen. CONCLUSION: Although
antinucleolar reactivity is more common in the first-degree relatives of patients
with SSc than controls, SSc associated ANoA are only present in patients with the
disease, and appear to be mutually exclusive.
=====================================================================
28.) [Systemic scleroderma and sarcoidosis: 3 new cases]
=====================================================================
Sclerodermie systemique et sarcoidose: trois nouveaux cas.
Bandt MD; Meyer O; Masson C; Peroux-Goumy L; Audran M; Kahn MF
Service de Rhumatologie, Hopital Bichat, Paris.
Ann Med Interne (Paris) (FRANCE) 1996 147 (8) p590-4 ISSN: 0003-410X
Language: FRENCH Summary Language: ENGLISH
Document Type:
JOURNAL ARTICLE; REVIEW; REVIEW OF REPORTED CASES English Abstract
Journal Announcement: 9707
Subfile: INDEX MEDICUS
We observed 3 patients with successive scleroderma (SS) and (what is considered to
be) sarcoidosis (SA). The diagnosis SS included in the 3 patients: Raynaud's
syndrome with pulpal necrosis and capillaritis, sclerodactylia and acro-osteolysis,
multiple joint pain and FAN+. Also observed were: esophagus involvement (n = 3),
pulmonary artery hypertension (n = 1), telangiectasia (n = 2) and anti-Scl 70 (n = 2).
Initially, all patients had restrictive pulmonary disease. SS was diagnosed 5 to 9
years prior to SA in 2 patients. Diagnosis of SA was based on the following
arguments: Loefgren's syndrome with erythema nodosa (n = 1), parotiditis (n = 2),
sicca syndrome (n = 2), myalgia (n = 2), joint involvement (n = 2), non-infectious
pluropericarditis (n = 2), epitheloid and giant cell granulomas without caseous
necrosis (lung = 3, liver = 1, lymph nodes = 1, salivary glands = 1, synovia = 1),
negative search for bacilli, elevated conversion enzyme (n = 1) and, in each case, by
the lack of any other cause. One patient died from lung cancer and another from
respiratory failure. Nome of the patients had primary biliary cirhosis. This rare
association between SS and SA demonstrates the confluent limits of certain systemic
diseases and raises a difficult problem to differentiate pulmonary involvement in
these diseases. The gravity of this localization and the poor sensitivy to
corticosteroids. (10 References)
=====================================================================
29.) [Cardiopathy in systemic sclerosis]
=====================================================================
La cardiopatia sclerodermica.
Valentini G; Maione S
Cartedra di Reumatologia, Seconda Universita, Napoli.
Recenti Prog Med (ITALY) Nov 1996 87 (11) p557-63 ISSN: 0034-1193
Language: ITALIAN Summary Language: ENGLISH
Document Type:
JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL English Abstract
Journal Announcement: 9706
Subfile: INDEX MEDICUS
Cardiac involvement is quite frequent in systemic sclerosis (SSc). From a
pathophysiologic point of view, one must differentiate a primary scleroderma heart
disease due to pericardial and/or myocardial and/or small coronary intramyocardial
vessel involvement from heart disease secondary to either pulmonary interstitial or
vascular involvement (pulmonal cor) or to kidney disease (hypertensive myocardial
disease). A significant difference emerges when the prevalence of clinically and
standard ECG detected cardiac involvement in SSc patients is compared with that
registered at autopsy. In the last years, however, Holter ECG, echocardiography,
perfusional scintigraphy and ventriculography have reduced such gap, a preclinical
scleroderma heart disease being detected by such techniques in quite a high
percentage of SSc patients. Asymptomatic SSc patients may be found to present small
pericardial effusions and/or either fixed (fibrosis) or reversible (vascular disease)
or both types thallium defects or a defective cardiac functional reserve. Both
clinically evident scleroderma heart disease and ventricular arrhythmias have a poor
prognostic significance. Therefore, a complete cardiological work-up must be
periodically carried out in SSc patients. Scleroderma heart disease has long been
considered a condition difficult to treat. The detection of diastolic abnormalities
and of diastolic failure in SSc patients make us able to understand the therapeutic
failure of inotropic agents. Recently, captopril has been shown to improve cardiac
function in SSc. It might act either on vascular disease or on fibrosis by affecting
the remodelling process of the myocardial wall. (40 References)
=====================================================================
30.) Scleroderma and pregnancy.
=====================================================================
Steen VD
Division of Rheumatology, Immunology, and Allergy, Georgetown University Medical
Center, Washington, DC, USA.
Rheum Dis Clin North Am (UNITED STATES) Feb 1997 23 (1) p133-47 ISSN: 0889-857X
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9706
Subfile: INDEX MEDICUS
Pregnancy in systemic sclerosis may be uneventful, with both good maternal and
fetal outcomes. Because scleroderma is a multisystem disease and complications do
occur, however, careful antenatal evaluations, discussion of potential problems, and
participation in a high-risk obstetric monitoring program is very important to
optimize the best outcome. Because women with diffuse scleroderma are at greater
risk for developing serious cardiopulmonary and renal problems early in the disease,
they should be encouraged to delay pregnancy until the disease stabilizes. All
patients who become pregnant during this high-risk time should be monitored extremely
carefully. Although there are some suggestions that there are increases in
infertility and miscarriages before disease onset, recent studies show that these
issues probably do not have major impact for women with established scleroderma who
plan to become pregnant. The high risk of premature and small infants may be
minimized with specialized obstetric and neonatal care, however. Renal crisis in
scleroderma is the only truly unique aspect of these pregnant, which, unlike blood
pressure elevation in nonscleroderma pregnancies, must be treated aggressively with
ACE inhibitors. Other pregnancy problems may not be unique to scleroderma, but
because it is a chronic illness, any complication carries higher risks for both
mother and child. Careful planning, close monitoring, and aggressive management
should allow women with scleroderma to have a high likelihood of a successful
pregnancy. (82 References)
=====================================================================
31.) Epidemiology of systemic sclerosis.
=====================================================================
Silman AJ; Newman J
University of Manchester, ARC Epidemiology Research Unit, School of Epidemiology
and Health Sciences, UK.
Curr Opin Rheumatol (UNITED STATES) Nov 1996 8 (6) p585-9 ISSN: 1040-8711
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9705
Subfile: INDEX MEDICUS
There have been few recent studies of the descriptive epidemiology of systemic
sclerosis, but in recent work the limited form of the disease seems more prominent
than reported in previous studies. Molecular genetic investigation of systemic
sclerosis remains disappointing in identifying susceptibility alleles. There are
some associations in relation to HLA class II alleles, specifically DP, DQ, and DR.
These associations, however, seem to be more important in predicting the nature of
the autoimmune response rather than describing disease susceptibility itself. The
study of occupational and environmental influences has been dominated by studies on
the role of silicone gel breast implants. These studies, driven by medicolegal
constraints, have overwhelmingly failed to prove any association. Other studies
confirm the continuing likelihood that organic solvents are implicated, at least in a
proportion of cases. (38 References)
=====================================================================
32.) Gastrointestinal features of scleroderma.
=====================================================================
Sjogren RW
Gastroenterology Section, Kaiser Permanente Medical Center, Fall Church, VA 22046,
USA.
Curr Opin Rheumatol (UNITED STATES) Nov 1996 8 (6) p569-75 ISSN: 1040-8711
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9705
Subfile: INDEX MEDICUS
Gastrointestinal involvement occurs in most patients with systemic sclerosis and is
subclinical in about one third. Early pathology is characterized by vasculopathy,
resulting in tissue ischemia and progressive dysfunction. Noninvasive esophageal
studies using semisolid bolus scintigraphy are sensitive but lack specificity. Long-
term treatment of reflux with high-dose proton pump inhibitors appears safe and
effective for symptom relief and may prevent recurrence of esophagitis and stricture.
Dyspepsia may result from gastroparesis and antral distension. Gastric antral
vascular ectasia is a vascular manifestation, and bleeding may be controlled
endoscopically. Prokinetic agents effective in pseudoobstruction include
metoclopramide, domperidone, cisapride, octreotide, and erythromycin. Patients with
intestinal neuropathy or response to bolus octreotide are more probable long-term
responders. The combination of octreotide and erythromycin may be particularly
effective in systemic sclerosis. The combination of cisapride and erythromycin may
cause serious cardiac arrhythmia and is contraindicated. Omeprazole may predispose
to small intestinal bacterial overgrowth. Malabsorption not responding to antibiotic
therapy should be investigated with small-bowel biopsy to rule out more unusual
causes. Pneumatosis cystoides intestinalis may be due to excessive hydrogen
production by intestinal bacteria altering the partial pressure of nitrogen in the
intestinal wall. In selected cases, surgery for intestinal failure is an option with
resection or bypass of affected segments or placement of enterostomy tubes for
feeding or decompression. Careful preoperative characterization of intestinal
segments is required. (57 References)
=====================================================================
33.) Systemic sclerosis in German uranium miners under special consideration of
autoantibody subsets and HLA class II alleles.
=====================================================================
Baur X; Rihs HP; Altmeyer P; Degens P; Conrad K; Mehlhorn J; Weber K; Wiebe V
Berufsgenossenschaftliches, Forschungsinstitut fur Arbeitsmedizin, Ruhr-Universitat
Bochum, Deutschland.
Respiration (SWITZERLAND) 1996 63 (6) p368-75 ISSN: 0025-7931
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9705
Subfile: INDEX MEDICUS
Systemic sclerosis (scleroderma) is a connective tissue disease with a wide range
of clinical manifestations, with high or low degrees of skin and internal organ
involvement together with different antinuclear antibody (ANA) specificities.
Several studies provide evidence that males, who are rarely affected by systemic
sclerosis, have an increased risk when working in mines. Therefore we reinvestigated
21 male subjects and 6 cases of deceased male patients who had been engaged in East
German uranium mines and had shown evidence of this disease in medical examinations.
Dermatological investigations, evaluation of chest X-rays and autoantibody estimation
were performed. PCR-sequence-specific oligonucleotide typing was used to study the
genetic association of HLA-D alleles with autoantibodies typical for scleroderma in
these uranium miners suffering from systemic sclerosis and in patients with
idiopathic systemic sclerosis. The determined HLA phenotype frequencies and the
following statistical analysis (Fisher's exact test (2-sided)) revealed that in
comparison with randomly selected controls, alleles DRB1*0300 (DR3) and DQB1*0201
(DQ2) were distinctly increased in the group of affected uranium miners, especially
in those with anti-Scl-70 positivity. In contrast, we did not observe significant
differences between affected and unaffected miners. Comparing anti-Scl-70-positive
affected uranium miners with anti-Scl-70-positive idiopathic systemic sclerosis cases.
DRB1*0300 as well as DQB1*0201 were also significantly enhanced in the former group.
ACA-positive systemic sclerosis miners had significantly elevated frequencies in
DRB1*0100 (DR1) and DRB1*0800 (DR8) only in comparison with unaffected miners and
unexposed controls. Our genetic and immunological data lead to the assumption that a
different set of HLA-D alleles in combination with exogenous factors is involved in
the induction of anti-Scl-70 antibodies in uranium miners that might influence their
susceptibility to the disease, whereas the same occupational exposure seems to have
no influence on the induction of ACA antibodies.
=====================================================================
34.) DNA allelic alterations within VNTR loci of scleroderma families.
=====================================================================
Artlett CM; Black CM; Briggs DC; Stephens C; Welsh KI
Oxford Transplant Centre, Churchill Hospital, London.
Br J Rheumatol (ENGLAND) Dec 1996 35 (12) p1216-22 ISSN: 0263-7103
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9704
Subfile: AIM; INDEX MEDICUS
We have characterized genetic alterations at the molecular level in 49 scleroderma
and 45 control families using variable number tandem repeats (VNTRs). Additionally,
paired fibroblast cell lines from the 'affected' and 'unaffected' skin and peripheral
blood leucocytes of 30 patients were also examined. All families in this study were
typed for Class I Cw alleles and Class II-DRB, -DQA and -DQB to confirm family
membership. There were significant rises in the level of VNTR mutations in
scleroderma patients (36.7%, n = 18), their siblings (16.3%, n = 13) and offspring
(21.7%, n = 15). The level of VNTR mutations in the control group was 0.6% (n = 5).
These mutations did not correlate with the presence of autoantibodies and no patient
was taking a known clastogenic drug. The most common VNTR site for mutation was
pYNZ22 (17p13.4). Differences were also seen in the VNTR alleles between fibroblast
and lymphocyte DNA from the same patient, as measured by size alteration of one of
the alleles. We have found that VNTRs are unstable in scleroderma patients,
relatives and offspring. The reason for the genomic changes remains unknown, but
previous studies have implicated the presence of a clastogen.
=====================================================================
35.) Treatment of systemic sclerosis.
=====================================================================
Pope JE
University of Western Ontario, London, Canada.
Rheum Dis Clin North Am (UNITED STATES) Nov 1996 22 (4) p893-907 ISSN: 0889-
857X
Language: ENGLISH
Document Type: JOURNAL ARTICLE; REVIEW; REVIEW, TUTORIAL
Journal Announcement: 9704
Subfile: INDEX MEDICUS
Guidelines for the conduct of clinical trials in progressive systemic sclerosis
have been recommended to determine drug efficacy better. To date, the results of
disease-modifying drugs in scleroderma have been disappointing. The treatment of
esophagitis has been revolutionized by omeprazole. Raynaud's phenomenon can be
treated with calcium channel blockers and iloprost. Scleroderma renal crisis can be
treated with aggressive blood pressure control using angiotensin converting enzyme
inhibitors. The best treatment for rapidly progressive scleroderma lung is still
unknown. Future treatments in scleroderma should be tested with the use of
recommended guidelines. (95 References)
=====================================================================
36.) Iloprost effects on phagocytes in patients suffering from ischaemic diseases: in
vivo evidence for down-regulation of alpha M beta 2 integrin.
=====================================================================
Mazzone A; Mazzucchelli I; Fossati G; Gritti D; Girola S; Canale C; Cusa C;
Ricevuti G
Department of Internal Medicine, University of Pavia, Italy.
Eur J Clin Invest (ENGLAND) Oct 1996 26 (10) p860-6 ISSN: 0014-2972
Language: ENGLISH
Document Type: CLINICAL TRIAL; JOURNAL ARTICLE
Journal Announcement: 9704
Subfile: INDEX MEDICUS
This study has been designed to demonstrate the in vivo effects of iloprost therapy
on expression of adhesion molecules on phagocytes. Sixty patients suffering from
peripheral arterial occlusive disease (PAOD) and/or from skin ulcers due to secondary
progressive systemic sclerosis (PSS) were enrolled in a double-blind controlled
parallel study. Thirty patients (group I) underwent iloprost infusion and 30
patients (group II) were treated with aspirin. Clinical assessment and measurement
of phagocyte activation in vivo, using quantitative flow cytometry, were performed on
entry and after 6 h on the first day of therapy. After 3 months of therapy, complete
healing of all cutaneous lesions was observed in 84% of the patients treated with
iloprost compared with the control patients (P < 0.001). Neutrophils and monocytes
of PAOD and PSS patients showed a significant decrease in the expression of the alpha
M beta 2 integrin adhesion receptor after 6 h of iloprost infusion. Neutrophils and
monocytes released a lower amount of anion superoxide (O2-) after 6 h of iloprost
treatment. These data confirm other clinical observations but demonstrate that in
vivo this drug modifies the expression of the alpha M beta 2 integrin of phagocytes
that has a key role in leukocyte-endothelium interactions in cases of inflammation
and thrombosis.
=====================================================================
37.) Association of esophagitis and esophageal strictures with diseases treated with
nonsteroidal anti-inflammatory drugs.
=====================================================================
El-Serag HB; Sonnenberg A
University of New Mexico, Albuquerque, USA.
Am J Gastroenterol (UNITED STATES) Jan 1997 92 (1) p52-6 ISSN: 0002-9270
Language: ENGLISH
Document Type: JOURNAL ARTICLE
Journal Announcement: 9704
Subfile: INDEX MEDICUS
BACKGROUND: It has been speculated that intake of nonsteroidal anti-inflammatory
drugs (NSAIDs) represents a risk factor for the occurrence of esophagitis and
esophageal strictures. METHODS: A case-control study was conducted to compare the
occurrence of comorbid diseases treated with NSAIDs in case and control subjects with
and without esophageal disease, respectively. The case population was comprised of
all patients with esophagitis (International Classification of Diseases code 530.1)
or esophageal stricture (code 530.3) who were discharged from hospitals of the
Department of Veteran Affairs between 1981 and 1994. In separate multivariate
logistic regressions, the occurrence of esophagitis or esophageal stricture served as
the outcome variable, and age, gender, ethnicity, and comorbid occurrence of an NSAID-
related diagnosis served as modifier variables. RESULTS: A total of 101,366
individual case subjects were included, of whom 92,860 presented with esophagitis and
14,201 with stricture. The occurrence of erosive esophagitis was associated with
osteoarthritis (odds ratio = 1.42, 95% confidence interval = 1.36-1.48), osteoporosis
(1.38, 1.25-1.52), back pain (1.49, 1.42-1.56), femur bone fracture (1.46, 0.92-
2.32), fibrositis (1.57, 1.41-1.75), tension headache (1.34, 1.27-1.40), ankylosing
spondylitis (1.33, 1.24-1.42), rheumatoid arthritis (1.13, 1.05-1.21), sicca syndrome
(1.15, 1.05-1.26), and systemic sclerosis (6.16, 4.65-8.14). NSAID-related diagnoses
represented similar risk factors for both esophagitis and esophageal stricture.
CONCLUSIONS: A large variety of diseases treated by NSAIDs are associated with a
significantly increased risk of esophageal erosion or stricture; the risk appears
similar for both of these. In some comorbid conditions, the underlying disease
process may contribute to the occurrence of esophageal pathology.
=====================================================================
DATA-MÉDICOS/DERMAGIC-EXPRESS No (56) 18/05/99 DR. JOSE LAPENTA R.
=====================================================================
Produced by Dr. José Lapenta R. Dermatologist
Venezuela
1.998-2.024
Producido por Dr. José Lapenta R. Dermatólogo Venezuela 1.998-2.0024
Tlf: 0414-2976087 - 04127766810












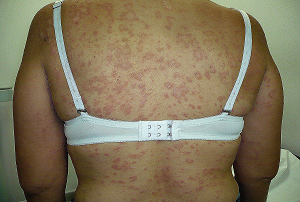




.png)